

the hepatitis B virus HBx protein in vivo and in vitro. Oncogene. 1998, 17, 2115-2123.
81.Zhu N., Khoshnan A., Schneider R., Matsumoto M., Dennert G., Ware C., Lai M.M. Hepatitis C virus core protein binds to cytoplasmic domain of tumor necrosis factor (TNF) receptor 1 and enhances TNFinduced apoptosis. J Virol. 1998, 72, 3691-3697.
82.O'Connell J. Fas ligand and the fate of antitumour cytotoxic T lymphocytes. Immunology. 2002, 105, 263-266.
83.Аббасова С.Г., Липкин В.М., Трапезников Н.Н., Кушлинский Н.Е. Система FAS-FASL в норме и патологии. Вопр биол мед фарм химии. 1999, 3, 3-17.
84.Rust C., Gores G.J. Apoptosis and liver disease. Am J Med. 2000, 108, 567-574.
85.Jo M., Kim T.H., Seol D.W., Esplen J.E., Dorko K., Billiar T.R., Strom S.C. Apoptosis induced in normal human hepatocytes by tumor necrosis factor-related apoptosis-induced ligand. Nat Med. 2000, 6, 564567.
86.Morgan M., Thorburn J., Pandolfi PP., Thorburn A. Nuclear and cytoplasmic shuttling of TRADD indyces apoptosis via different mechanisms. J Cell Biol. 2002, 157, 975-984.
87.Kmiec Z. Cooperation of liver cells in health and disease. Adv Anat Embryol Cell Biol. 2001, 161, 1-151.
88.Jaeschke H., Smith C.W. Mechanism of neutrophyl-induced parenchymal cell injury. J Leukoc Biol. 1997, 61, 647-653.
89.Lawson J.A., Farhood A., Hopper R.D., Bajt M.L., Jaeschke H. The hepatic inflammatory response after acetaminophen overdose. Role of neutrophils. Toxicol Sci. 2000, 54, 509-516.
90.Olson T.S., Ley K. Chemokines and chemokine receptors in leukocyte trafficking. Am J Physiol Regul Integr Comp Physiol. 2002, 283, 7-28.
91.McClain C.J., Song Z., Barve S.S., Hill D.B. Deaciuc I. Recent advances in alcoholic liver disease. IV. Dysregulated cytokine metabolism in alcoholic liver disease. Am J Physiol Gastrointest Liver Physiol. 2004, 287, 497-502.
92.Bone-Larson C.L., Simpson K.J., Colletti L.M., Lukacs N.W, Chen S.C., Lira S., Kunkel S.L., Hogaboam C.M. The role of chemokines in the immunopathology of liver. Immunol Rev. 2000, 177, 8-20.
93.Bautista A.P. Impact of alcohol on the ability of Kupffer cells to produce chemokines and its role in alcoholic liver disease. J Gastroenterol Hepatol. 2000, 15, 349-356.
94.Boisvert J., Kunkel E.J., Campbell J.J., Keeffe E.B., Butcher E.C., Greenberg H.B. Liver-infiltrating lymphocytes in end-stage hepatitis C virus: subsets, activation status, and chemokine receptor phenotypes. J
191
Hepatol. 2003, 38, 67-75.
95.Jaeshke H. Cellular adhesive molecules: regulation and functional significance in the pathogenesis of liver diseases. Am J Physiol Gastrointest Liver Physiol. 1997, 273, 602-611.
96.Essani N.A., Bajt M.L., Vondervecht S.L., Farhood A., Jaeschke H. Transcriptional activation of vascular ell adhesive molecule-1 (VCAM-1) gene in vivo and its role in the pathophysiology of neu- trophil-induced liver injury in murine endotoxin shock. J immunol. 1997, 158, 5941-5948.
97.Patel T., Gores G., Kaufmann S.H. The role of proteases during apoptosis. FASEB J. 1996, 10, 587-597.
98.Shibuya H., Ohkohchi N., Tsukamoto S., Satomi S. Tumor necrosis factor-induced, superoxide-mediated neutrophil accumulation in cold ischemic/reperfused rat liver. Hepatology. 1997, 26, 113-120.
99.Lemasters J.J. V. Necrapoptosis and mitochondrial permeability transition: shared pathways to necrosis and apoptosis. Am J Physiol. 1999, 276, 1-6.
100.Ziol M., Tepper M., Lohez M., Arcangeli G., Ganne N., Christidis C., Trinchet J.C., Beaugrand M., Guillet J.G., Guettier C. Clinical and biological relevance of hepatocyte apoptosis in alcoholic hepatitis. J Hepatol. 2001, 34, 254-260.
101.Losser M.R., Payen D. Mechanisms of liver damage. Semin Liver Dis. 1996, 16, 357-367.
102.Ивашкин В.Т., Шульпекова Ю.О. Неалкогольный стеатогепатит. РМЖ. Болезни органов пищеварения. 2000, 2, 41-45.
103.James O., Day C.P. Non-alcoholic steatohepatitis (NASH): a disease of emerging identity and importance. J Hepatol. 1998, 29, 495-501.
104.Pessayre D., Berson A., Fromenty B., Mansouri A. Mitochondria in steatohepatitis. Semin Liver Dis. 2001, 21, 57-69.
105.Никитин И. Г., Сторожаков Г. И. Лекарственные поражения печени. РЛС. Болезни печени и желчевыводящих путей. М. 2002, Из-во "Издательский дом".
106.Шерлок Ш., Дули Дж. Заболевания печени и желчных путей. Медицина, М. 1999, 540-548.
107.Mari M., Wu D., Nieto N., Cederbaum A.L. CYP2E1-depend- ent toxicity and up-regulation of antioxidant genes. J Biomed. 2001, 8, 52-58.
108.Liu H., Jones B.E., Bradham C., Czaja M. Increased cytochrome P-450 2E1 expression sensitizes hepatocytes to c-Jun-mediated cell death from TNF-alpha. Am J Physiol Gastrointest Liver Physiol. 2002, 282, 257-266.
109.Muriel P. Regulation of nitric oxide synthesis in the liver. J Appl Toxicol. 2000, 20, 189-195.
192
110.Gumpricht E., Dahl R., Yerushalmi B., Devereaux M.V., Sokol R.J. Nitric oxide ameliorates hydrophobic bile acid-induced apoptosis in isolated rat hepatocytes bynon-mitochondrial pathways. J Biol Chem. 2002, 277, 25823-25830.
111.Kurose I., Miura S., Higuchi Y., Watanabe N., Kamegaya Y., Takaishi M., Tomita K., Fukumura D., Kato S., Ishii H. Increased nitric oxide synthase activity as a cause of mitochondrial dysfunction in rat hepatocytes: roles for tumor necrosis factor alpha. Hepatology, 1996, 24, 1185-1192.
112.Machida K., Cheng K.T., Sung V.M., Lee K.J., Levine A.M., Lai M.M. Hepatitis C virus infection activates the immunologic (type II) isoform of nitric oxide synthase and thereby enhances DNA damage and mutations of cellular genes. J Virol. 2004, 78, 8835-8843.
113.Meadows M. Serious liver injury: Leading reason for drug removals, restrictions. FDA Consum. 2001, 35, 8-9.
114.Bissell D.M., Cores G.J., Laskin D.L., Hoofnage J.H. Druginduced liver injury: mechanism and test systems. Hepatology. 2001, 33, 1009-1013.
115.Morgan E.T. Regulation of cytochrome p450 by inflammatory mediators: why and how? Drug Metab Dispos. 2001, 29, 207-212.
116.Chin K., Kong A.N. Application of DNA microarrays in pharmacogenomics and toxicogenomics. Pharm Res. 2002, 19, 1773-1778.
117.Thomson R.K., Arthur M.J. Mechanisms of liver cell damage and repair. Eur J Gastroenterol Hepatol. 1999, 11, 949-955.
118.Riordan S.M., Williams R. Mechanisms of hepatocyte injury, multiorgan failure, and prognostic criteria in acute liver failure. Semin
Liver Dis. 2003, 23, 203-215.
119.Ding W.X., Yin X.M. Dissection of the multiple mechanisms of TNF-alpha-induced apoptosis in liver injury. J Cell Mol Med. 2004, 8, 445-454.
120.Шагидулин М.Ю, Противоишемическая защита печеночного трансплантанта с помощью липина - водорастворимого препарата липидной природы. Автореферат дисс. канд. М. 2005 г.
193
6. МЕМБРАННЫЕ ФЕРМЕНТНЫЕ СИСТЕМЫ: ИХ ПОВРЕЖДЕНИЕ И РЕПАРАЦИЯ
6.1. Инактивация ферментных систем биомембран.
Повреждения биомембран и сопряженных с ними функций большинства интегральных и примембранных белков, включая рецепторы, транспортные и структурообразующие белки, ферменты и ферментные системы, как плазматической мембраны, так и мембран различных субклеточных органелл, нарушают течение многих клеточных процессов и функционирование клетки в целом. В последние годы появляется все больше данных, доказывающих, что эффекты ПОЛ лежат в основе многих повреждающих механизмов, включая повреждение мембран и встроенных в них ферментных систем. Интерес к этим процессам особенно возрос в связи с выяснением их роли в патогенезе многих заболеваний [1].
Обработка мембранных препаратов in vitro реагентами, удаляющими или повреждающими мембранные фосфолипиды (экстракция органическими растворителями, дезоксихолатом или действие фосфолипаз) приводит к резкой инактивации мембранносвязанных ферментов. In vivo под действием повреждающих факторов происходит активация ПОЛ. Взаимодействие продуктов ПОЛ с белковыми структурами приводит к инактивации мембранных ферментов и ферментных комплексов, таких как глюкозо-6-фосфатаза, цитохром Р450, транспортные АТФ-азы, аденилатциклаза, переносчики электронов
дыхательной цепи митохондрий и др. [2]. Наиболее чувствительны к перекисному повреждению мембранные ферменты, содержащие SН-группы в активном центре, в первую очередь Ca2+-АТФазы. Их инактивация играет большую роль в патологии клетки, так как вызывает замедление "оттока" ионов кальция из клетки, выравнивание концентраций ионов Са2+ во внутри- и межклеточном пространстве, нарушение внутриклеточного гомеостаза и развитие повреждений внутриклеточных и плазматической мембран (см. главу 4).
В зависимости от области поражения механизмы повреждения мембранных ферментов можно условно разделить на две группы:
(1) повреждение самого ферментного белка и (2) повреждение микроокружения фермента, влияющего на активность последнего.
Помимо свободнорадикального окисления, в последние годы в качестве причины повреждения мембранных ферментных белков отмечают также гликирование (например, при сахарном диабете) [3]. При этом в организме в качестве компенсаторной реакции может наблюдаться нарушение регуляции экспрессии генов
194
ферментов и соответственное изменение их количества. В частности, это показано для Са2+-АТФазы саркоплазматического ретикулума мышечной ткани при алкогольной миопатии. Патологическое увеличение как количества, так и активности фермента связывают с компенсаторной реакцией на индуцированные алкоголем мембранные повреждения, вызванные образованием аддуктов мембранных белков с ацетальдегидом [4]. Нарушение регуляции экспрессии генов ферментов (с соответствующим повышением синтеза) показано и для примембранных ("заякоренных") сериновых протеаз в условиях опухолевого роста. В нормальных клетках активность этих ферментов не обнаруживается [5].
Повреждение мембранных белков наблюдали в клетках миокарда при ишемии и реперфузии. При этом происходило разобщение особенно важных для сократительной деятельности миокарда Са2+- è Na+,K+-АТФаз - в результате как аноксии, так и действия супероксидов, свободных радикалов, и пероксинитритов, образующихся при взаимодействии супероксидов с оксидом азота [6].
Âнастоящее время воздействие АФК считают универсальным механизмом повреждения как белковых, так и липидных компонентов мембраны [7] (см. главу 4). Особенно существенным является в этом отношении повреждающее действие легко
диффундирующей молекулы Н2Î2. Окислительные повреждения мембранных ферментов суммируются с ингибирующим действием на их активность изменений физико-химических свойств липидного слоя мембран (включая аннулярные липиды), которые
происходят за счет процессов ПОЛ и воздействия эндогенных фосфолипаз [7, 8] (см. главу 4).
Âоснове нарушения активности мембранных ферментов при повреждении фосфолипидного бислоя лежат различные механизмы. Они зависят от сложности строения фермента, его локализации в мембране, характера катализируемой реакции и др. Так, для ферментов, превращающих гидрофобные субстраты, фосфолипидный бислой является необходимой средой для реакции и его изменения (например, усиление ригидности под действием ПОЛ), существенно влияют на эффективность ферментативных реакций [9]. Для олигомерных ферментов мембранная липидная матрица выполняет роль "организатора" субъединиц, поэтому ее повреждение нарушает пространственное взаиморасположение протомеров и координацию отдельных звеньев ферментативного процесса. Это показано, например, для микросомальной глюкозо-6-фосфатазы, состоящей из каталитического и транспортного белков, для проявления
195
активности которых необходимы ФХ и ФЭ соответственно; гидролиз этих ФЛ фосфолипазами инактивирует данный фермент [10].
Классическим примером мембранного липид-зависимого фермента является Na+,K+-АТФаза, хотя требования к липидному окружению для нее не столь специфичны и ограничиваются необходимостью обеспечения амфифильности окружения и низкой микровязкости [11]. Этот фермент включает как водорастворимые, так и трансмембранные субъединицы, которые при повреждениях мембраны разобщаются, инактивируя тем самым фермент.
Нарушение структурной организация липидного бислоя может также затруднять взаимодействие фермента с субстратами, находящимися в водной фазе [9]. Возможно и прямое воздействие поврежденного участка мембраны на фермент, приводящее к изменению положения активного центра и его доступности для субстрата.
Этот факт особенно важен для таких интегральных мембранных белков-ферменов, как β-глюкоцереброзидаза, УДФглюкуронилтрансфераза, глюкозо-6-фосфатаза, стеароил-КоА- десатураза и ряда других [12].
Аналогичным воздействиям при повреждениях мембран подвергаются и другие мембранные белки, участвующие в трансмембранном переносе веществ и лиганд-рецепторных взаимодействиях, что приводит к нарушению функции клеток.
Некоторые исследователи отмечают возможность адаптации
клетки, включая работу мембранных белков, к таким повреждениям за счет реакций, осуществляемых PK C, МАРК и митохондриальными АТФ-зависимыми кальциевыми каналами. Открытие этих каналов, например, вызывает деполяризацию митохондрий, снижая избыточное накопление Са2+ при реперфузии, что способствует регенерации клеток миокарда [13]. В случае глубоко зашедшего процесса нарушение активности мембранных ферментов может вызвать такие изменения клеточного метаболизма, которые приведут к некротической или апоптотической гибели клетки и развитию патологии в целом.
В последние годы особое внимание уделяется роли процессов внутриклеточной сигнализации в регуляции активности некоторых мембранных ферментов. Это показано, в частности, для ключевого фермента процессов коагуляции крови - трансмембранного гликопротеина, называемого "тканевым фактором". Этот фермент обнаружен в адвентиции кровеносных сосудов и во внутренней области атеросклеротической бляшки.
196
Тканевый фактор, экспрессированный на мембранах моноцитов и макрофагов, может участвовать в воспалительных процессах и в дестабилизации бляшки. Отмечают его чувствительность к ингибиторам, синтезируемым эндотелиальными клетками сосудов
âпроцессе сигналинга [14].
Âбольшинстве работ, опубликованных в основном в 80х-90-х годах прошлого века, показана возможность восстановления активности поврежденных мембранных ферментов. Например, добавление ФХ или смеси ФХ и лизоФХ к препаратам микросом печени крысы,
обработанных in vitro фосфолипазами А или С, восстанавливало активность Са2+-АТФазы, Na+,K+-АТФазы, глюкозо-6-фосфатазы, цитохром b5-редуктазы, УДФ-глюкуронилтрансферазы, стеароил-
КоА-десатуразы, липазы, фенилаланингидроксилазы; подобные результаты получены и для сфингомиелиназы мозга, β-оксибутират- дегидрогеназы митохондрий сердца быка, ацетилхолинэстеразы эритроцитов [15, 16].
Âбольшинстве случаев восстановление фосфолипидами активности мембранных ферментов, по-видимому, носит неспецифический характер, т.е. для большинства ферментов не существует специфической потребности в определенном типе ФЛ, и репарирующую функцию могут выполнять разные типы ФЛ и их смеси [17]. Для активности же некоторых мембранных ферментов оказался важным жирнокислотный состав добавляемых ФЛ, что было показано для Са2+-АТФазы саркоплазматического ретикулума [18]. Авторы получали частично очищенную АТФазу в комплексе с фосфолипидами, а затем модифицировали входящие в состав
комплекса ФЛ. Показано, что замена собственных ФЛ на ДПФХ снижала активность в 8 раз по сравнению с частично делипидированным ферментом, а при замене на диолеоил-ФХ скорость реакции, наоборот, возрастала. Другие авторы также отмечают существенное требование к высокой подвижности углеводородных цепей для восстановления активности мембранных ферментов, а в ряде случаев - к наличию определенного поверхностного заряда [19].
Âэкспериментах in vivo была показана возможность восстановления мембран клеток печени с помощью диеты, содержащей ненасыщенные ФЛ. Так, диета с растительным ФХ предотвращает развитие у животных холестаза, вызванного
циклоспорином А; его действие связано с ингибированием АТФаз (Na+,K+-, Ca2+-, Mg2+-) [20]. Подобные результаты получены и для других, менее изученных мембранных ферментов. Например, отмечается активирующий эффект кислых фосфолипидов (ФС, ФИ и ФК) на миозиновую фосфатазу ткани гортани [21]. Показана
197
способность анионных ФЛ (особенно ФС) активировать 5- монодегидрогеназу плаценты человека [22], а также делипидированную малатдегидрогеназу микобактерий. В последнем случае образуется активный комплекс фермента со смесью кардиолипина и ФЭ [23]. Сообщается также об активирующем действии анионных ФЛ на периферические макрофаги [24]. На добавление фосфолипидов реагирует и сопряженная с G-белком рецепторная киназа [25].
Таким образом, повреждения мембран вызывают изменения в активности мембранных ферментов, что приводит к нарушению клеточных функций и развитию патологии. Добавление фосфолипидов, как показано в ряде работ, способствует восстанавлению активности мембранных ферментов.
6.2. Инактивация ферментных систем мембран ЭР как показатель повреждения печени и их репарация фосфолипидами.
В многочисленных исследованиях показано повреждающее действие различных токсических агентов на мембраны ЭР, в первую очередь, клеток печени, и защитное действие ФХ на поврежденные мембраны, в частности на цитохром Р450 - ключевой фермент монооксигеназной системы печени, участвующей в детоксикационных и биосинтетических процессах. Изучение молекулярных механизмов повреждения мембран ЭР, взаимосвязи повреждения фосфолипидного компонента и сопутствующей инактивации мембранных ферментов является важнейшей предпосылкой для поиска факторов, способствующих восстановлению структуры и
функции поврежденных мембран [26 - 31]
Êнастоящему времени накоплено много данных, свидетельствующих
îтом, что повреждения мембран ЭР печени, сопровождающиеся инактивацией цитохрома Р450 и одновременным нарушением процессов гидроксилирования экзогенных (лекарства, яды, пищевые добавки, канцерогены и т. д.) и эндогенных субстратов (стероиды, холестерин, простагландины, жирные кислоты), вызывают нарушение функций печени человека и животных [29, 32-35].
Монооксигеназная система печени оказалась чувствительной к действию целого ряда токсических агентов, таких как четыреххлористый
углерод (ССl4), гелиотрин, тиоацетамид, алкилирующие соединения, радиация и т. д. [29, 36-39].
Хроническое введение животным ССl4 широко используется в качестве модели при исследованиях развития патологических изменений в печени. Особенности проявления действия этого яда, давно заинтересовавшие исследователей, заключаются в его высокой гепатотоксичности. Известно, что ключевым или решающим в повреждающем эффекте ССl4 является
198
воздействие его метаболитов, вероятнее всего, свободных радикалов, как наиболее реакционноспособных молекул. Основным механизмом повреждающего действия этого яда на печень является ускоренное образование перекисей липидов. Процессы превращения ССl4 протекают в мембранах ЭР печени в присутствии НАДФН [29, 38]. Установлено, что введенный экспериментальным животным ССl4 обнаруживается во всех мембранах печеночной клетки, но наибольшее его количество находится в мембранах ЭР. ССl4 связывается с цитохромом Р450, как с апоферментом, так и с железом гема, по типу СО. Спектральная константа связывания ССl4 с гидрофобным участком составляет 0,5 мМ, а с железом - 0,4 мМ [40].
Связывание яда с цитохромом Р450 имеет решающее значение в механизме его повреждающего действия, так как ССl4 в присутствии НАДФН восстанавливается до трихлорметилового радикала (ССl•3), который играет роль инициатора процессов перекисного окисления ПНЖК в мембранах. Повреждение самого цитохрома Р450 происходит за счет двух взаимодополняющих механизмов: (1) непосредственного повреждения белка ССl•3 радикалами и (2) повреждения его липидного окружения в мембране ЭР [31, 39]. Считают, что образованный на цитохроме Р450 радикал ССl•3 может атаковать двойные связи ненасыщенных жирных кислот ("метиленовые мостики"), присоединять атом водорода, переходя при этом в инертную молекулу хлороформа (СНСl3) и генерируя липидные радикалы; последние, инициируя цепь процессов перекисного окисления липидов, повреждают мембраны (см. главу 4) [37].
О восстановлении ССl4 цитохромом Р450 с участием НАДФНзависимой цепи переноса электронов свидетельствуют многочисленные
экспериментальные данные. Так, было показано, что специфичные ингибиторы цитохрома Р450, такие как цианид натрия или SKF-525 А (β,β- диэтиламиноэтилдифенилпропилацетат), оказывают угнетающее действие на скорость превращения ССl4 в препаратах микросом и в опытах наживотных[37,41].Такжебылообнаружено,чтоживотные,находящиеся на малобелковой диете, а также новорожденные крысята и эмбрионы человека, в печени которых снижено содержание цитохрома Р450, обладают низкой чувствительностью к действию ССl4 [42]. В то же время, индукция НАДФН-специфичной системы метаболизма и увеличение экспрессии цитохрома Р450 при введении фенобарбитала и бензпирена, резко усиливают токсическое действие ССl4 [29, 36, 37].
Прооксидантное действие радикалов, образующихся из ССl4, было экспериментально подтверждено при исследовании скорости накопления малонового диальдегида в НАДФН- и аскорбат-зависимых системах перекисного окисления ПНЖК в микросомах. Введение в инкубационную смесь тетрахлорметана сопровождалось появлением, в случае НАДФНзависимой системы ПОЛ, перекисных продуктов вследствие резкого
199
усиления реакций перекисного окисления ПНЖК ССl•3 радикалами [32, 33, 35].
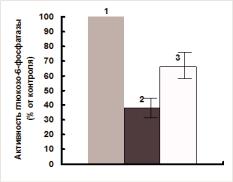
Таким образом, одним из важнейших эффектов ССl4 является прооксидантное действие его метаболитов и интенсификация процессов ПОЛ. В печени уже в ранние сроки после введения яда обнаруживаются продукты реакций перекисного окисления и резкое снижение содержания арахидоновой и декозогексаеновой кислот в sn-2 положении основных фосфолипидных компонентов мембран эндоплазматического ретикулума - ФХ и ФЭ. Оказалось также, что даже небольшого снижения содержания этих двух ненасыщенных кислот (на 8-10%) достаточно для выраженного (50%-ного) ингибирования другого микросомального мембранного фермента - глюкозо-6-фосфатазы [43]. Показано, что этот фермент теряет свою активность под влиянием ССl4 êàê in vitro, òàê è in vivo [44], и этот факт считают следствием нарушения структуры гидрофобных участков мембран в процессе перекисного окисления ненасыщенных жирных кислот ФЛ, которое может быть частично восстановлено путем инкубации микросом с фосфолипидами. Так, на рис. 6.2.1 показано, что активность глюкозо-6-фосфатазы в печени крыс, отравленных ССl4, снижена и составляет ~ 25% от активности фермента у контрольных животных. Инкубация микросом печени отравленных животных с фосфолипидами (ФХ) восстанавливает активность этого фермента, но не полностью, а ~ до 70% от контроля [27, 28, 32, 38, 45].
Рис. 6.2.1. Активность глюкозо-6-ôосфатазы микросом печени крыс, отравленных ССI4, и ее восстановление после инкубации
in vitro с фосфолипидами
Крысам внутрибрюшинно однократно вводили CCl4 в дозе 0,1 мл/100 г массы. Через 60 мин аналогично вводили фосфолипиды в виде липосом в дозе 10 мг/100 г массы. Через 24 часа животных забивали, выделяли фракцию микросом печени. В микросомах определяли активность глюкозо-6-фосфатазы, в сравнении с таковой в микросомах здоровых животных [45]. 1 - контроль; 2 - введение CCl4; 3 - введение CCl4 и фосфолипидов в виде липосом.
200