

activation and its role in cardiac cell death. Circ Res. 2003, 92, 589-591.
98.Santana P., Pena L.A., Haimovitz-Friedman A., Martin S., Green D., McLoughlin M., Cordon-Cardo C., Schuchman E.H., Fuks Z., Kolesnick R. Acid sphingomyelinase-deficient human lymphoblasts and mice are defective in radiation-induced apoptosis. Cell. 1996, 86, 189–199.
99.Pena L.A., Fuks Z., Kolesnick R.N. Radiation-induced apoptosis of endothelial cells in the murine central nervous system: protection by fibroblast growth factor and sphingomyelinase deficiency. Cancer Res. 2000, 60, 321–332.
100.Paris F., Fuks Z., Kang A., Capodieci P., Juan G., Ehleiter D., Haimovitz-Friedman A., Cordon-Cardo C., Kolesnick R. Endothelial apoptosis as the primary lesion initiating intestinal radiation damage in mice. Science. 2001, 293, 293–297.
101.Haimovitz-Friedman A., Cordon-Cardo C., Bayoumy S., Garzotto M., McLoughlin M., Gallily R., Edwards C.K., Schuchman E.H., Fuks Z., Kolesnick R. Lipopolysaccharide induces disseminated endothelial apoptosis requiring ceramide generation. J Exp Med. 1997, 186, 1831–1841.
102.Andrieu-Abadie N., Jaffrezou J.P., Hatem S., Laurent G., Levade T., Mercadier J.J. L-Carnitine prevents doxorubicin-induced apoptosis of cardiac myocytes: role of inhibition of ceramide generation. FASEB J. 1999, 13, 1501–1510.
103.Lawlor M., Alessi D. PKB/Akt: a key mediator of cell proliferation, survival and insulin responses? J Cell Sci. 2001, 114, 2903-2910.
104.Harada-Shiba M., Kinoshita M., Kamido H., Shimokado K. Oxidized low density lipoprotein induces apoptosis in cultured human umbilical vein endothelial cells by common and unique mechanisms. J Biol Chem. 1998, 273, 9681–9687.
105.Deigner H.P., Claus R., Bonaterra G.A., Gehrke C., Bibak N., Blaess M., Cantz M., Metz J., Kinscherf R. Ceramide induces aSMase expression: implications for oxLDL-induced apoptosis. FASEB J. 2001, 15, 807–814.
106.Xia P., Wang L., Gamble J.R., Vadas M.A. Activation of sphingosine kinase by TNF inhibits apoptosis in human endothelial cells. J Biol Chem. 1999, 274, 34499–34505.
107.Cuvillier O., Pirianov G., Kleuser B., Vanek P.J., Coso O.A., Gutkind J.S., Spiegel S. Suppression of ceramide-mediated programmed cell death by sphingosine-1-phosphate. Nature. 1996, 381, 800–803.
108.Hisano N., Yatomi Y., Satoh K., Akimoto S., Mitsumata M., Fujino M.A., Ozaki Y. Induction and suppression of endothelial cell apoptosis by sphingolipids: a possible in vitro model for cell-cell interactions between platelets and endothelial cells. Blood. 1999, 93, 4293–4299.
109.Lee M.J., Thangada S., Claffey K.P., Ancellin N, Liu C.H., Kluk M., Volpi M., Sha’afi R.I., Hla T. Vascular endothelial cell adherens junction assembly and morphogenesis induced by sphingosine-1-phos- phate. Cell. 1999, 99, 301–312.
111
110.Kwon Y.G., Min J.K., Kim K.M., Lee D.J., Billiar T.R., Kim Y.M. Sphingosine-1-phosphate protects human umbilical vein endothelial cells from serum-deprived apoptosis by nitric oxide production. J Biol Chem. 2001, 276, 10627–10633.
111.Malagarie-Cazenave S., Andrieu-Abadie N., Segui B., Gouaze V., Tardy C., Cuvillier O., Levade T. Sphingolipid signalling: molecular basis and role in TNF - induced cell death. Expert Rev Mol Med. 2002, 20, 1-15.
112.Henis Y.I., Moustakas A., Lin H.Y., Lodish H.F. The types II and III transforming growth factor-beta receptors form homooligomers. J Cell Biol. 1994, 126, 139-154.
113.Gilboa L., Wells R.G., Lodish H.F., Henis Y.I. Oligomeric structure of type I and type II transforming growth factor beta receptors: homodimers form in the ER and persist at the plasma membrane. J Cell Biol. 1998, 140, 767-777.
114.Yanaga F., Watson S.P. TNF-alpha stimulates sphingomyelinase through the 55 kDa receptor in HL-60 cells. FEBS Lett. 1992, 314, 297300.
115.Modur V., Zimmerman G.A., Prescott S.M., McIntyre T.M. Endothelial cell inflammatory responses to TNF. J Biol Chem. 1996, 271, 13094–13102
116.Kim I., Moon S.O., Kim S.H., Kim H.J., Koh Y.S., Koh G.Y. Vascular endothelial growth factor expression of intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1), and E-selectin through nuclear factor-B. J Biol Chem. 2001, 276, 7614–7620.
117.Slowik M.R., De Luca L.G., Min W., Pober J.S. Ceramide is not a signal for TNF-induced gene expression but does cause programmed cell death in human vascular endothelial cells. Circ Res. 1996, 79,
736–747.
118.Xia P., Gamble J.R., Rye K.A., Wang L., Hii C.S., Cockerill P., Khew-Goodall Y., Bert A.G., Barter P.J., Vadas M.A. TNFα induces adhesion molecule expression through the sphingosine kinase pathway. Proc Natl Acad Sci USA. 1998, 95, 14196–14201.
119.Xia P., Wang L., Gamble J.R., Vadas M.A. Activation of sphingosine kinase by TNFα inhibits apoptosis in human endothelian cell. J Biol Chem. 1999, 274, 34499-34505.
120.Wang A., Dennis E.A. Mammalian lysophospholipases. Biochim Biophys Acta. 1999, 1439, 1-16.
121.Yatomi Y., Ohmori T., Rile G., Kazama F., Okamoto H., Sano T., Satoh K., Kume S., Tigyi G., Igarashi Y., Ozaki Y. Sphingosine-1- phosphate as a major bioactive lysophospholipid that is released from platelets and interacts with endothelial cells. Blood. 2000, 96, 3431–3438.
122.English D., Welch Z., Kovala A.T., Harvey K., Volpert O.V., Brindley D.N., Garcia J.G. Sphingosine-1-phosphate released from platelets during clotting accounts for the potent endothelial cell chemotactic activity of blood serum and provides a novel link between hemo-
112
stasis and angiogenesis. FASEB J. 2000, 14, 2255–2265.
123.Lee O.H., Kim Y.M., Lee Y.M., Moon E.J., Lee D.J., Kim J.H., Kim K.W., Kwon Y.G. Sphingosine-1-phosphate induces angiogenesis: its angiogenic action and signaling mechanism in human umbilical vein endothelial cells. Biochem Biophys Res Commun. 1999, 264, 743–750.
124.Wang F., Van Brocklyn J.R., Hobson J.P., Movafagh S., Zukowska-Grojec Z., Milstien S., Spiegel S. Sphingosine-1-phosphate stimulates cell migration through a Gi-coupled cell surface receptor. J Biol Chem. 1999, 274, 35343–35350.
125.Olivera A., Spiegel S. Sphingosine-1-phosphate as second messenger in cell proliferation induced by PDGF and FCS mitogens. Nature. 1993, 365, 557-560.
126.Sano T., Baker D., Virag T., Wada A., Yatomi Y., Kobayashi T., Igarashi Y., Tigyi G. Multiple mechanisms linked to Platelet Activation result in Lysophosphatidic Acid and Sphingosine 1-Phosphate generation in blood. J Biol Chem. 2002, 227, 21197–21206.
127.Pyne S., Pyne N.J. Sphingosine-1-phosphate signalling in mammalian cells. Biochem J. 2000, 349, 385–402.
128.Hla T., Lee M.J., Ancellin N., Liu C.H., Thangada S., Thompson B.D., Kluk M. Sphingosine-1-phosphate: extracellular mediator or intracellular second messenger? Biochem Pharmacol. 1999, 58, 201–207.
129.Spiegel S., Milstein S. Sphingosine-1-phosphate, a key cell signaling molecule. J Biol Chem. 2002, 277, 25851-25854.
130.Sachinidis A., Kettenhofen R., Seewald S., Gouni-Berthold I., Schmitz U., Seul C., Ko Y., Vetter H. Evidence that lipoproteins are carriers of bioactive factors. Arterioscler Thromb Vasc Biol. 1999, 19, 2412-2421.
131.Gouni-Berthold I., Sachinidis A. Does the coronart risk factor low density lipoprotein alter growth and signaling in vascular smooth muscle cells? FASEB J. 2002, 16, 1477-1488.
132.Ghosh T.K., Bian J., Gill D.L. Intracellular calcium release mediated by sphingosine derivatives generated in cells. Science. 1990, 248, 1653–1656.
133.van Koppen C.J., Meyer Zu Heringdorf D., Laser K.T., Zhang C., Jakobs K.H. Bunemann M., Pott L. Activation of a high affinity Gi protein-coupled plasma membrane receptor by sphingosine-1-phos- phate. J Biol Chem. 1996, 271, 2082–2087.
134.Nakajima N., Cavalli A.L., Biral D., Glembotski C.C., McDonough P.M., Ho P.D., Betto R., Sandona D., Palade P.T., Dettbarn C.A., Klepper R.E., Sabbadini R.A. Expression and characterization of Edg-1 receptors in rat cardiomyocytes: calcium deregulation in response to sphingosine-1-phosphate. Eur J Biochem. 2000, 267, 5679–5686.
135.Goetzl E.J., An S. Diversity of cellular receptors and functions for the lysophospholipid growth factors lysophosphatidic acid and sphingosine 1-phosphate. FASEB J. 1998, 12, 1589-1598.
136.Chun J., Goetzl E.J., Hla T., Igarashi Y., Lynch K.R., Moolenaar
113
W., Pyne S., Tigyi G. International Union of Pharmacology. XXXIV. Lysophospholipid receptor nomenclature. Pharmacol Rev. 2002, 54, 265-269.
137.Проказова Н.В., Звездина Н.Д., Коротаева А.А. Влияние лизофосфатидилхолина на передачу трансмембранного сигнала внутрь клетки. Биохимия. 1998, 63, 38-46.
138.Silliman C.C., Elzi D.J., Ambruso D.R., Musters R.J., Hamiel C., Harbeck R.J., Paterson A.J., Bjornsen A.J., Wyman T.H., Kelher M., England K.M., McLaughlin-Malaxecheberria N., Barnett C.C., Aiboshi J., Bannerjee A. Lysophosphatidylcholines prime the NADPH oxidase and stimulate multiple neutrophil functions through changes in cytosolic calcium. J Leucoc Biol. 2003, 73, 511-524.
139.Hunter G., Bigelow D., Squier T. Lysophosphatidylcholine modulates catalytically important motions of Ca-ATPase phosphorylation domain. Biochemistry. 1999, 38, 4604-4612.
140.Fang X., Gaudette D., Furui T., Mao M., Estrella V., Eder A., Pustilnik T., Sasagawa T., Lapushin R., Yu S., Jaffe R.B., Wiener J.R., Erickson J.R., Mills G.B. Lysophospholipid growth factors in the initiation, progression, metastases, and management of ovarian cancer. Ann N Y Acad Sci. 2000, 905, 188-208.
141.Budnik L.T., Mukhopadhyay A.K.. Lisophosphatidic acid and its role in reproduction. Biol Reprod. 2002, 66, 859-865.
142.Tokumura A., Miyake M., Nishioka Y., Yamano S., Aono T., Fukuzawa K. Production of lysophosphatidic acids by lysophospholipase D in human follicular fluids of In vitro fertilization patients. Biol Reprod. 1999, 61, 195-199.
143.Galaria I., Fegley A.J., Nicholl S.M., Roztocil E., Davies M.G. Differential regulation of ERK1/2 and p38(MAPK) by components of the Rho signaling pathway during sphingosine-1-phosphate-induced smooth muscle cell migration. J Surg Res. 2004, 122, 173-179.
144.Santos W.L., Rossi J.A., Boggs S.D., MacDonald T.L. The molecular pharmacology of lysophosphatide signaling. Ann N Y Acad Sci. 2000, 905,233–241.
145.Sorensen S.D., Nicole O., Peavy R.D., Montoya L.M., Lee C.J., Murphy T.J., Traynelis S.F., Hepler J.R. Common signaling pathways link activation of murine PAR-1, LPA, and S1P receptors to proliferation of astrocytes. Mol Pharmacol. 2003, 64, 1199-1209.
114
ГЛАВА 4. РОЛЬ ФОСФОЛИПИДОВ В ПРОЦЕССАХ ПОВРЕЖДЕНИЯ КЛЕТКИ
К настоящему времени накоплено много данных о роли фосфолипидов в структурной организации и функционировании клетки. Это важно для изучения и понимания молекулярных механизмов возникновения и развития патологий, многие из которых, как правило, сопровождаются изменениями внутриклеточных органелл, структурной организации и целостности биомембран и, в той или иной степени, повреждениями клетки в целом. Нарушение функций одних клеток может быть первопричиной развития болезни, а создавшиеся неблагоприятные изменения в организме влекут за собой нарушение функционирования клеток других тканей и дальнейшее развитие патологического процесса. Например, гибель клеток сердечной мышцы при инфаркте миокарда может привести к гипоксии и нарушению функций клеток других органов: почек, мозга, печени [1-5].
Процессы, приводящие к повреждению клетки, давно привлекали внимание исследователей. В середине прошлого века основным критерием повреждения было повышение проницаемости клеточной мембраны [6,7]. Позднее, (в 70х80х годах), с усовершенствованием методов выделения, анализа и конструирования мембран, было получено множество данных, свидетельствующих о первичности мембранных нарушений едва ли не во всех видах клеточных повреждений [8]. В последние годы развитие таких направлений современной биохимии, как геномика
èпротеомика, позволило глубже понять многие молекулярные процессы, протекающие в клетке, в том числе и механизмы ее повреждения [9]. Одновременный мониторинг многих типов молекул дал возможность изучать клеточные ответы на молекулярные повреждения, включая эволюционно закрепленные системы индуцируемой молекулярной защиты. Все это позволило выявить новые биомаркеры, действия которых основаны на молекулярных механизмах клеточного ответа на повреждения [9].
Âмногочисленных работах, опубликованных в последние годы
èсвязанных с поведением клетки в различных повреждающих условиях (как in vitro, так и in vivo, включая патологическое состояние организма и внешние воздействия), повреждения мембранных систем клетки занимают существенное место либо как первая повреждаемая мишень, либо как система, определяющая во многом дальнейшую судьбу клетки [1, 2, 10].
115
4.1.Основные изменения в клетках при повреждении
4.1.1.Реакция клетки на изменения условий.
Âусловиях нормального гомеостаза существование клетки ограничено довольно узким диапазоном функциональных и структурных характеристик, определяемых ее генетической программой метаболизма, дифференциации и специализации, окружением соседних клеток, а также доступностью метаболических субстратов. При некотором изменении условий или каких-либо патологических стимулах возможен ряд физиологических и морфологических изменений, при которых достигается новое стационарное состояние, защищающее жизнеспособность клетки и вносящее некоторые изменения в ее функции в ответ на внешние стимулы. Такое состояние называют адаптацией клеток. Она, например, может включать обогащение мембранных фосфолипидов ПНЖК при пониженных температурах
[11].Это препятствует снижению жидкостности мембраны, или снижению экспрессии генов, кодирующих SREBP белки, участвующие в биосинтезе холестерина в ответ на его избыточное поступление в клетку [12, 13] и т.д.
Если адаптационные возможности клетки превышены, возникает последовательность событий, называемая повреждением. До какого-то момента повреждение может быть обратимым, но если воздействие повреждающего фактора оказывается длительным или усиливается, повреждение становится необратимым, и клетка гибнет. Реакция клетки на
повреждение зависит как от характера повреждающего стимула, так и от свойств самой клетки. Так, клетки с высокой скоростью метаболизма (нейроны, клетки сердечной мышцы) в наибольшей степени подвержены повреждениям (ишемия при гипоксических воздействиях) [14]. Кроме того, подверженность клеток и тканей (особенно печени) повреждающему фактору может зависеть от состояния или особенностей организма. Так, голодание повышает
гепатотоксичность СС14, а индивидуальная непереносимость к агентам (например, к алкоголю или каким-либо лекарствам) усиливает их повреждающее действие [15]. Любой патологический процесс, какой бы степенью функциональных нарушений он не сопровождался, начинается на уровне ультраструктур или биохимических систем клетки, и только после достижения некоторого уровня их дезорганизации в поврежденной клетке начинают проявляться морфологические изменения.
4.1.2.Причины, вызывающие повреждение клетки.
Все причины, вызывающие повреждение клетки и связанные с
116
воздействием как на организм в целом, так и непосредственно на клетки, можно условно разделить на несколько основных категорий.
1)Недостаток кислорода, например, при ишемии, сердечной или дыхательной недостаточности, анемии, или отравлении окисью углерода.
2)Физические воздействия, к которым относятся механическая травма, резкое изменение температуры, внезапные изменения атмосферного давления, радиация, электрический шок, и т.д.
3)Химические агенты, к которым относят: (а) обычные субстраты типа глюкозы или соли, но в повышенных (гипертонических) количествах; (б) токсические соединения, в том числе тяжелые металлы, растворители, яды, гормоны, (в) лекарства
èäð.
4)Инфекционные агенты (вирусы, бактерии);
5)Анафилактические и аутоиммунные реакции (и измененные иммунные ответы вообще).
6)Генетические нарушения как в отдельных генах, так и аномалии в хромосомах.
7)Питательный дисбаланс, связанный с недостаточностью (или, наоборот, с избыточностью) поступления в организм белков, витаминов.
8)Старение, действие которого может проявляться или специфически, или в комбинации с выше названными факторами.
Структурные и биохимические ассоциации в клетке настолько взаимосвязаны, что каким бы ни был локус первоначальной
повреждающей атаки, она все равно приводит к широкому ряду вторичных эффектов.
4.2.Апоптоз и некроз - два варианта гибели поврежденной клетки.
Морфологические различия физиологической и патологической гибели клетки впервые подробно были описаны более 50 лет назад [16], а термин "апоптоз", как обозначение формы гибели клетки, радикально отличающейся от некроза, прочно укоренился в 70-х годах прошлого века [17]. С тех пор считается общепризнанным существование двух основных форм клеточной гибели, одинаковых по результату (прекращение биологической активности клетки и фагоцитоз ее остатков), но отличающихся по молекулярно-биохимическим, морфологическим и клиническим критериям.
К морфологическим критериям апоптоза относятся целостность плазматической мембраны, конденсация хроматина, набухание
117
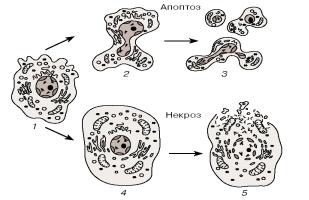
митохондриальной мембраны. Остатки клетки фагоцитируются макрофагами или соседними клетками, поэтому клеточное содержимое не попадает в межклеточное пространство и не вызывает воспалительной реакции. При некрозе, напротив, клетки набухают, их митохондрии и другие органеллы увеличиваются в объеме, разрываются внутриклеточные и плазматическая мембраны. В результате активируются лизосомальные ферменты, а внутриклеточное содержимое, попадая во внеклеточную среду, вызывает воспалительные процессы (рис.4.2.1) [4, 18, 19 ].
Рис. 4.2.1. Морфологические изменения клетки при апоптозе и некрозе (по [22] с изменениями)
1 - нормальная клетка, 2 - апоптотическое сморщивание клетки с образованием пузырчатых выростов, 3 - фрагментация клетки с образованием апоптотических телец, 4 - набухание клетки при некрозе, 5 - некротическая дезинтеграция клетки.
Путь гибели, "выбираемый" повреждаемой клеткой, зависит от многих факторов, включая тип клетки, ее энергетический статус (т.е. уровень синтеза АТФ), характер и степень повреждения, состояние иммунной системы организма, в частности активности клеток Т-киллеров, и др. [20, 21].
4.2.1. Апоптоз.
Апоптоз - процесс "запрограммированной гибели клеток", который в здоровом организме сбалансирован их физиологической регенерацией [23]. В ходе апоптоза внутренние или внешние факторы, активируя посредством передачи сигналов генетическую программу, вызывают гибель клетки и ее эффективное удаление из ткани [20, 21]. По выражению Скулачева В.П. [24], "Апоптоз служит молекулярным
118
механизмом, обеспечивающим старение и смерть живого организма, что, возможно, не очень полезно для индивида, но чрезвычайно важно для стабильности популяции". Ежедневно около 5% клеток в организме подвергаются апоптозу, исчезая бесследно в течение 15 - 120 минут, а их место занимают новые [25]. Таким образом, апоптоз - генетически контролируемый биологический механизм, ответственный за поддержание постоянства численности и выбраковку дефектных клеток.
Среди молекулярно-биохимических признаков апоптоза важнейшими являются:
(1)Энергозависимость. В отличие от некроза, апоптоз требует обязательного наличия АТФ, изменение его уровня может определить направление клеточной гибели. По мнению некоторых исследователей, решающим может являться даже не абсолютное содержание АТФ, а отношение АТФ/АДФ в клетке [26].
(2)Расщепление ядерной ДНК с формированием крупных фрагментов, кратных, как правило, 180-200 пар нуклеотидов, что приводит к формированию ДНКовой "лестницы" на электрофореграмме;
(3)Уменьшение объема клетки. На ранних стадиях апоптоза клетка сморщивается, теряя до 1/3 своего объема за несколько минут.
(4)Активация специфических цистеиновых протеаз (каспаз) и, соответственно, деградация белка в клетке.
(5)Появление (экстернализация) фосфатидилсерина на наружной поверхности плазматической мембраны клетки и
образующихся из нее апоптотических телец, что способствует их узнаванию макрофагами и фагоцитозу.
Апоптоз является результатом действия различных внешних и внутренних факторов. Неспецифические внешние факторы способны индуцировать апоптоз, который при увеличении воздействия повреждающего агента может перейти в некроз клетки
[27].К физиологическим факторам апоптоза относят как внутриклеточные сигналы, так и внешние, опосредующие свое действие через рецепторные системы. Среди них наибольший интерес представляют гормоны. Они могут выступать как индукторами, так и ингибиторами гибели клетки, в зависимости от стадии ее дифференцировки (например, половые гормоны) [28, 29].
Другим физиологическим регулятором апоптоза являются цитокины, представляющие обширную группу продуцируемых клетками полифункциональных сигнальных белков, действующих при связывании со специфическими рецепторами на клеткахмишенях [30]. В зависимости от структуры и функции цитокины
119
подразделяют на три большие группы: ростовые факторы, семейство TNF и спиральные цитокины (интерлейкины, интерфероны). Эффект цитокинов на клетки неоднозначен: для одних клеток они выступают в роли индукторов, для других - ингибиторов апоптоза. Это зависит от типа клетки, стадии ее дифференцировки и фукционального состояния и т.д. [31]. Одновременное присутствие в клетке индукторов и ингибиторов апоптоза свидетельствует о том, что программируемая гибель клетки напрямую зависит от соотношения этих регуляторов.
Различают несколько фаз апоптоза или программированной гибели клетки: (1) инициация апоптоза; (2) трансдукция (проведение) апоптотического сигнала; (3) активация каспаз; (4) активация эндонуклеаз и специфическая деградация ДНК, которая
èприводит к гибели клетки.
4.2.2.Инициация и трансдукция апоптотического сигнала.
Âзависимости от стимулов, инициирующих апоптоз, можно выделить два главных внутриклеточных апоптозных сигнальных каскада: митохондриальный и рецепторный, опосредованный специфическими "рецепторами смерти" плазматической мембраны. Последние являются частью суперсемейства рецепторов TNF. Последовательность событий после стимуляции апоптоза наиболее изучена в настоящее время для рецептора Fas (Fas-R), также называемого CD95 [20, 25, 32]. Цитоплазматический домен Fas-R обогащен цистеином, а интегральная (мембранная) часть, содержит домен смерти DD, который вовлекается в белок-
белковое взаимодействие, генерируя сигнал смерти. Лиганд этого рецептора (Fas-L) - цитокин из семейства белков TNF, экспрессирующийся, главным образом, на активированных Т- лимфоцитах и естественных киллерах. Связывание лиганда Fas-L с рецептором (Fas-R/Fas-L) приводит к олигомеризации последнего и формированию сигнального комплекса, инициирующего каскад реакций, вызывающих апоптоз. В олигомеризации Fas участвуют:
(1) цитоплазматический DD-домен рецептора, (2) "адапторный" белок FADD (Fas-ассоциированный DD, содержащий DED - эффекторный домен смерти) и (3) прокаспазы-8. Образование комплекса этих белков активирует каспазу-8, и развивается характерный для апоптоза каскад активационных процессов [32]. Интересно, что у некоторых опухолевых клеток активность каспазы-8 может быть ингибирована специальным FLIP (FLICE inhibitory protein; FLICE - FADD-Like Interleukin Converting Enzyme) белком, который, связываясь с сигнальным комплексом, препятствует апоптозу [33]. Установлено, что мутации в генах Fas
120