

Конфигурационное взаимодействие
Сравнение расчетных величин с соответствующими экспериментальными значениями показывает сильные различия:
r*эксп<r*расчиEэксп>Eрасч.
Эти расхождения можно в значительной мере исправить, если применить специальный прием — учесть т.н. "конфигурационное взаимодействие" (КВ). Разновидность метода МО, в которой такой учет производится, называется методом МО-КВ.
С формальной точки зрения, учет КВ производится следующим образом: если у молекулы имеется несколько возможных состояний, описываемых волновыми функциями, у которых спиновой множитель один и тот же, а пространственные множители имеют один и тот же тип пространственной симметрии, то из этих волновых функций следует составить линейную комбинацию. При правильном подборе коэффициентов этой ЛК можно существенно улучшить расчетное значение полной энергии молекулы.
В случае молекулы водорода имеются две подходящие волновые функции: Ф1= [GG](–) иФ4= [UU](–), у которых спиновые множители полностью совпадают, а пространственные множители относятся к четному типу. Тогда исправленная с учетом КВ волновая функция молекулы будет иметь вид:
Фg = {C1[GG] + C2[UU]}( – )
(коэффициенты С1иС2можно оптимизировать вариационным методом).
С физической точки зрения это означает, что два чистых состояния — [GG] и [UU] — не являются стационарными и могут переходить друг в друга, а стационарным является только суперпозиционное состояниеФg.
Принципиальный момент заключается в том, что любому чистому состоянию соответствует строго определенная электронная конфигурация (т.е. способ распределения электронов по молекулярным орбиталям). Для суперпозиционного состояния указать конкретную конфигурацию невозможно. При попытке экспериментально установить положение электронной пары мы будем находить ее либо в конфигурации GG(с вероятностью |C1|2), либо в конфигурацииUU(с вероятностью |C2|2). Другими словами, истинное распределение электронов не может быть задано какой-либо одной конфигурацией — всегда необходимо учитывать несколько возможных вариантов. (Отсюда и термин — "конфигурационное взаимодействие".)
Можно проанализировать физический смысл конфигурационного взаимодействия и с другой стороны. Когда мы для представления молекулярных орбиталей пользуются атомным базисом (МО ЛКАО), делается серьезная ошибка. Настоящие базисные состояния определяются измерительным прибором — системой перегородок, разделяющих молекулу на одноядерные фрагменты. Результатом разделения является переход молекулярного одноэлектронного облака, делокализованного по всему ядерному остову молекулы, в облако, локализованное в окрестности только одного ядра. Именно такие конечные состояния и описывают правильные базисные функции 1,1, …,n. Хотя такая функция описывает электронное облако, локализованное в окрестности одного ядра, это облако не является полностью изолированным. Напротив, оно подвергается кулоновскому воздействию со стороны остальных достаточно близко расположенных ядер молекулы. Поэтому использование в качестве базисных функций атомных орбиталей, характерных для изолированных атомов, является довольно грубым приближением. Например, в изолированном атоме водорода состояние электрона точно описывается волновой функцией 1s, чему соответствует сферически симметричное распределение электронной плотности. Если к атому приблизить с одной стороны еще одно атомное ядро, то под его влиянием электронное облако деформируется, распределение электронной плотности уже не будет сферически симметричным, а волновая функция будет существенно отличаться от идеального типа 1s.
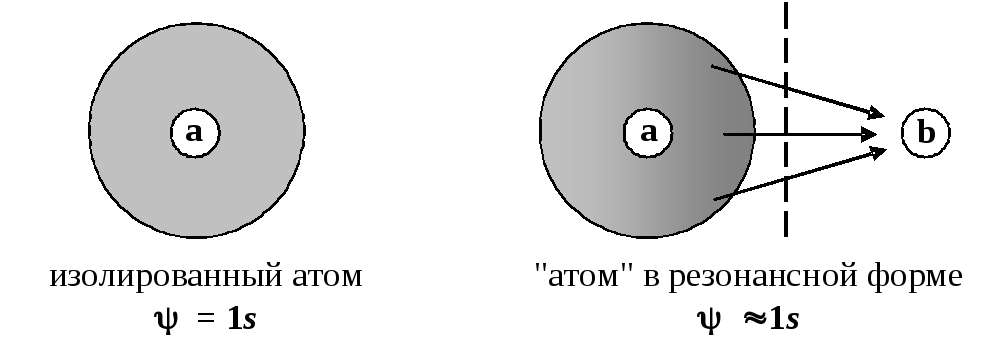
Включение в линейную комбинацию нескольких волновых функций для разных конфигураций приводит к более адекватной картине распределения электронной плотности в молекуле и, соответственно, к более низкой полной энергии.