

Влияние межъядерного расстояния
Энергии всех состояний молекулы зависят от расстояния между ядрами rab, которое входит в выражения для кулоновского и обменного интегралов, а также интеграла перекрывания. Проведя множество расчетов для серии межъядерных расстояний, можно построить зависимостьE=f(rab), график которой имеет следующий качественный вид:
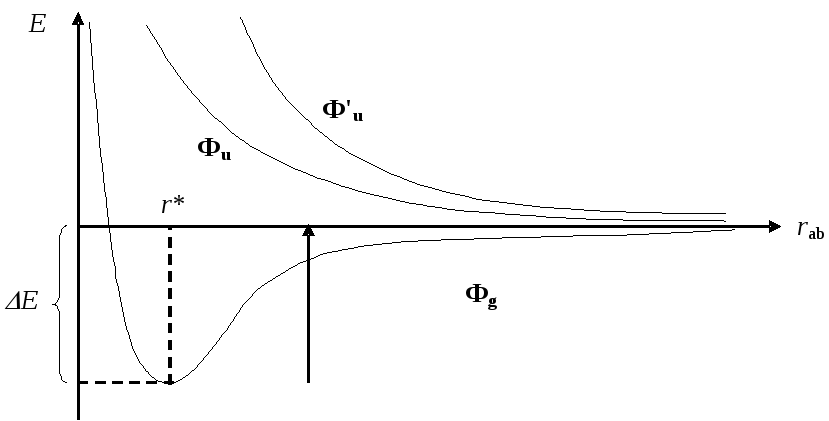
Из графика ясно видно, что процесс сближения атомов может приводить к двум принципиально разным результатам. Первый (Фg) — возникновениесил притяжения, что сопровождается уменьшением полной энергии системы по мере сближения атомов. Второй (ФuиФ'u) — возникновениесил отталкивания, что сопровождается возрастанием полной энергии системы. Ясно, что в первом случае сближение атомов будет происходить самопроизвольно до достижения состояния устойчивого равновесия с минимальной энергией (rab=r*). Именно этот случай соответствует образованию химической связи между атомами.
Следует подчеркнуть, что зависимости типа E=f(rab) могут быть полезными при решении ряда химических задач, связанных с оценкой реакционной способности молекул. Так, глубина минимумаЕна графике зависимостиE=f(rab) может служить для оценкипрочности химической связи, поскольку она соответствует порции энергии, необходимой для разделения молекулы на отдельные атомы. Кроме того, выделенное значение межъядерного расстоянияr*, соответствующее минимуму полной энергии, может служить для оценкидлины химической связи.
Подобные зависимости можно получать и для сложных, многоатомных молекул. В этом случае они изображаются многомерными функциями вида E=f(r12,r13,r23, … ,rij, …), графики которых называютсяповерхностями потенциальной энергии(ППЭ) молекулы. Располагая сведениями от виде ППЭ, можно решать многие важные проблемы, касающиеся химического строения молекул (установление цепи химического действия, оценки длин и энергий химических связей, наличие структурных изомеров) и химических превращений с их участием (оценки энергетических эффектов и энергий активации).
Полезно также обратить внимание на то, что образование химической связи сопровождается спариванием электронов атомов (состояние Фg синглетное), тогда как возникновение сил отталкивания наблюдается для триплетного состоянияФu, когда электроны не спарены. Можно сказать, что спаривание валентных электронов является необходимым признаком химического взаимодействия атомов. Этот признак, однако, недостаточен, поскольку в состоянииФ'uэлектроны спарены, но химического взаимодействия не наблюдается. Причина такого различия заключается в различной форме электронного облака. Если сравнить пространственные части двух синглетных волновых функций:
Фg = AB + BA и Ф'u = AA – BB
то обнаружится, что во втором случае в электронном облаке имеется разрыв (узловая поверхность, проходящая между ядрами):
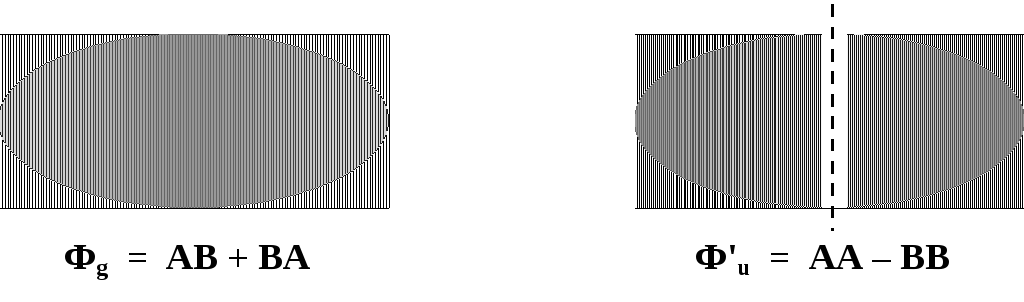
Следовательно, для образования химической связи кроме спаривания необходимо еще одно условие — надлежащее распределение плотности электронного облака в пространстве.
2.1.3. Общая формулировка метода вс
Процедура, рассмотренная выше в приложении к молекуле водорода, может быть легко обобщена на любые молекулы. В общем виде эта процедура включает в себя следующие этапы.
1. Определение набора РФ (всех возможных способов распределения электронов молекулы по "атомам").
2. Составление для всех "атомов" одноатомных волновых функций и их оптимизация, например, методом ССП ХФ.
3. Построение волновых функций РФ в виде произведений одноатомных волновых функций.
4. Анализ перестановочной и пространственной симметрии полученных произведений и построение линейных комбинаций, удовлетворяющих требованиям симметрии.
5. Оптимизация неопределенных коэффициентов на основе вариационного принципа.
6. Вычисление физических свойств молекулы, например, энергии.
Дополнительно можно отметить, что оператор Гамильтона для произвольной молекулы имеет вид (нерелятивистская модель):
H=(i) +(Ui) +(Uij) +(U)
где i= (–2/2m)2i— оператор кинетической энергииi-го электрона,
Ui= –Ze2/ri— оператор потенциальной энергииi-го электрона в кулоновском поле-го ядра,
Uij=e2/rij— операторы межэлектронного отталкивания,
U=Z Ze2/r— операторы межъядерного отталкивания.
Эта форма гамильтониана приводит к тому, что выражение для полной энергии молекулы приобретает вид:
E=E+(JK)
где Е— полная энергия изолированного атома с номером,
J— межатомные кулоновские интегралы (энергии кулоновского отталкивания атомов),
K— межатомные обменные интегралы (поправки к энергии за счет перехода электронов от одного атома к другому.
Заметим, что выражения для Есами имеют подобный вид:
E = Ei + (Jij Kij)
где Еi— остовные энергии электронов в-ом изолированном атоме,
Jij— внутриатомные межэлектронные кулоновские интегралы,
Kij— внутриатомные межэлектронные обменные интегралы.
Таким образом, в основе метода ВС лежит вспомогательная структурная модель, заимствованная из классической теории химического строения, согласно которой молекула представляет собой механический агрегат из индивидуальных атомов, свойства которых, однако, изменены за счет межатомных взаимодействий кулоновской природы. Поэтому метод ВС позволяет сопоставить вводимые в его рамках поправки на межатомное взаимодействие (J— межатомные кулоновские интегралы иK— межатомные обменные интегралы) обычным химическим связям, которым в классической химии также приписывается локализованный бинарный характер. Это позволяет получать некоторые количественные оценки таких характеристик химических связей, как длины, энергии диссоциации и др.