

1.5.6. Показания к цитогенетическому исследованию:
А. Исследование Х- и У - хроматина:
Неясен пол новорожденного;
Пренатально при подозрении на болезнь, передаваемую сцеплено с полом;
Первичная аменорея у женщин (при подозрении на 45, ХО или 46, ХУ);
Ранняя вторичная аменорея;
Бесплодие у мужчины (подозрение на 47, ХХУ).
Б. Исследование кариотипа:
Все лица с патологией Х - и У - хроматина;
Дети с множественными пороками развития (МПР);
При недифференцированной олигофрении;
Семьи с повторными мертворождениями и спонтанными выкидышами;
Родители детей с множественными пороками развития и при установлении у ребенка диагноза хромосомной болезни;
Сибсы, планирующие рождение детей, если в семье диагностирован случай хромосомной аберрации;
Пренатально у плода, если возраст матери более 35 лет.
Хромосомные болезни, обусловленные аномалиями аутосом.
1.5.7. Синдром или болезнь Дауна (трисомия 21)
Самая распространенная из хромосомных болезней. В общей популяции частота синдрома составляет 1 : 600 - 800 живорожденных. Однако болезнь в 2 раза чаще встречается у плодов, так как больше половины их при трисомии 21 абортируется на ранних сроках беременности.
Причина трисомии 21 - простое нерасхождение хромосом в мейозе. В настоящее время можно определить донора добавочной 21 хромосомы, им в 80% случаев является женщина, в 20% - мужчина. В 60% нерасхождение происходит в 1 фазе мейоза, т.е. когда вредный фактор действует на яйцеклетки плода (синдром “бабушки”); и в 40% - во вторую фазу мейоза, т.е. во время зачатия и первые дни после оплодотворения. Возраст матери влияет на нерасхождение хромосом во 2 фазу.
Формы синдрома Дауна:
Простая трисомия 21. Кариотип больного 47, ХУ (ХХ), + 21.
Эта форма составляет 95% всех случаев синдрома Дауна.
Транслокационная форма. Кариотип больного 46, ХУ (ХХ), -14, -21, +t (14q21q), 46, XX, -21, +t(21q21q). Эта форма составляет 4% всех случаев. Основная часть транслокаций относится к центрическому соединению 21 с 13, 14, 15 хромосомой, реже с 21 и 22. Транслокационная форма может быть результатом мутации, вновь возникшей в гаметах или зиготе, либо она унаследована от одного из родителей, который является фенотипически здоровым носителем (кариотип 45, ХХ (ХУ), -14, -21, +t (14q21q)).
Хромосомный мозаицизм. Кариотип больного 46, ХХ / 47, ХХ + 21. Данная форма составляет 1% всех случаев синдрома Дауна. Возникает на ранних стадиях дробления зиготы. Чем позднее наступило нерасхождение хромосом, тем меньше доля патологического клеточного клона. Если это происходит на поздних стадиях, клинические проявления будут скудными, но клетка из аномального клона может вступить в мейоз и послужить причиной рождения ребенка с синдромом Дауна.
Фенотипическая характеристика.
Характерна умственная отсталость, от полной идиотии до легкой дебильности.
Черепно - лицевые дисморфии: уплощенный затылок, монголоидный разрез глаз, эпикант, депигментированные пятна на роговице (пятна Брашфилда), высунутый язык, оттопыренные ушные раковины измененной формы, гипертелоризм, уплощенная переносица.
Грудная клетка: врожденные пороки сердца, в основном дефекты перегородки.
Органы брюшной полости и таза: уменьшенный ацетабулярный и подвздошный углы, малый размер полового члена, крипторхизм.
Руки и ноги: обезьянья складка, короткие широкие ладони, гипоплазия средней фаланги 5 пальца, щель между 1 и 2 пальцами.
Другие признаки: “готическое небо, косоглазие, короткая широкая шея, маленькие зубы, сморщенный язык, атрезия кишечника, неперфорированный анус.
Дерматоглифика: ульнарные петли на большинстве пальцев, радиальные петли на 4 и 5 пальцах, дистальный осевой трирадиус, большой угол atd, единственная складка на 5 пальце.
Расчет генетического риска при синдроме Дауна.
Эмпирический риск:
Простая трисомная форма :
а) суммарный риск зависит от возраста матери. До 20 лет - 0,08%; до 25 лет - 0,1%; 25-35 лет - 0,2%; 35-40 лет - 0,54%; свыше 40 лет -1,6%.
Б) повторный риск для сибсов больного в зависимости от возраста матери. До 35 лет - 1%; свыше 35 лет - удвоенный риск для данной возрастной группы.
Транслокационная форма:
а) риск для сибсов в спорадических случаях (при нормальном кариотипе родителей) - соответствует простой трисомии;
б) Риск для потомства носителей при семейных робертсоновских транслокациях -
|
Тип транслокации |
Пол женщина |
носителя, % мужчина |
|
(21q22q) |
7 |
2 |
|
(21qDq) |
10 |
2.4 |
|
(21q21q) |
100 |
100 |
Мозаицизм у родителей - риск для потомства определяется по формуле
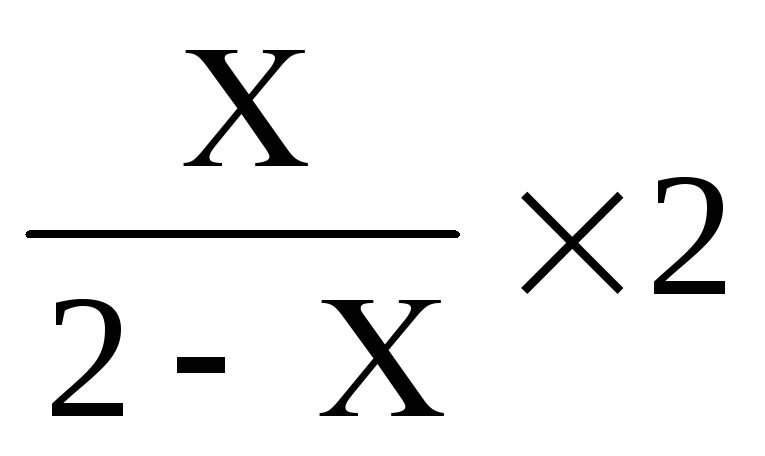 ;
где
х - доля аномального клеточного клона.
;
где
х - доля аномального клеточного клона.