

Ч 2
.pdfТаблица 7.1. Частота отдельных врождённых аномалий
Заболевание |
Частота при рождении |
Тип наследования |
|
|
|
Наследственные |
заболевания |
|
Ахондропластическая карликовость |
1/10 000 |
АД |
Муковисцидоз |
1/2000, белые (США) |
АР |
Галактоземия |
1/30 000–1/40 000 |
АР |
Гемофилия А |
1/2500, мужчины |
XР |
Семейная гиперхолестеринемия |
1/500 |
АД |
Серповидноклеточная анемия |
1/625, афроамериканцы |
АР |
Болезнь Тея–Сакса |
1/3600, евреи (ашкенази) |
АР |
Нейрофиброматоз |
1/3000 |
АД |
Хромосомные аномалии |
|
|
Синдром Клайнфельтера |
1/500, мужчины |
|
Синдром Тернера |
1/10 000, женщины |
|
Синдром Дауна |
1/800 |
|
Врождённые недоразвития |
|
|
Волчья пасть |
1/2000 |
|
Заячья губа |
1/1150 |
|
Косолапость (без пороков нервной системы) |
1/400 |
|
Врождённый вывих бедра (без пороков нервной |
|
|
системы) |
1/400 |
|
Недоразвитие конечностей |
1/2500 |
|
Расщелина – неполное срастание позвоночника |
|
|
(без анэнцефалии, т. е. отсутствия всего или |
1/2000 |
|
большей части мозга) |
|
|
Пороки сердца |
1/200 |
|
|
|
|
Примечание. АД, АР, XР – соответственно аутосомно-доминантный, аутосомно-рецес- сивный, X-сцепленный рецессивный типы наследования.
признака, обычно исчезающего к моменту рождения. И только к XIX в., когда причины врождённых аномалий были в основном уже изучены, суеверия прошлого заменили научные знания, а в XX в. (после развития генетики) были открыты тонкие механизмы возникновения аномалий у человека.
Причины возникновения врождённых аномалий (пороков). К дородо-
вым причинам пороков относятся наследственные факторы и/или воздействие окружающей среды на развитие зародыша. Причиной возникновения пороков во время родов могут быть травмы или инфекции. Как врождённый порок рассматривается также очень маленький вес при рождении, который отражает либо недоношенность, либо недостаточность процессов развития плода и является основной причиной детской смертности и инвалидности.
Причиной врождённых пороков развития могут быть мутации генов (мономутантные врождённые пороки), на долю которых приходится 14–17 %, хромосомные и геномные мутации (9–10 %), воздействие неблагоприятных
211
наследственных и средовых факторов (40–65%). Действие последних в эмбриогенезе приводит к формированию множественных врождённых пороков развития, частота которых составляет 0,9–2,4 случая на 1000 новорождённых. Чаще всего при синдроме множественных врождённых пороков развития поражаются сердечно-сосудистая (15 % по сравнению с 0,5–0,8 % в общей популяции) и мочеполовая (52 % против 10 % в популяции) системы. Однако возможно сочетание пороков сердца и почек с врождёнными пороками других органов.
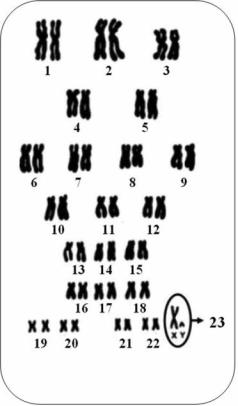
1. Наследственность. Ряд врождённых аномалий наследуется так же, как другие признаки. Информация от родителей детям передаётся с помощью генов (главными носителями последних служат хромосомы). У каждого человека в норме в половой клетке (сперматозоиде или яйцеклетке) находится 23 хромосомы. При оплодотворении формируется стандартный генетический набор из 46 хромосом. 22 из 23 хромосом репродуктивной клетки – аутосомы, т. е. они не определяют пол, а одна (X либо Y) является половой хромосомой
(рис. 7.1). Сперматозоид несёт X- и |
Y-хромосомы, яйцеклетка – только |
|
X-хромосомы. Оплодотворение яйце- |
|
клетки сперматозоидом с Y-хромо- |
|
сомой формирует организм мужского |
|
пола, с X-хромосомой – женского. |
|
Большинство наследственных при- |
|
знаков и их нарушения соответствуют |
|
статистически предсказуемым типам |
|
наследования, называемым менделев- |
|
скими (в честь их первооткрывателя |
|
Грегора Менделя (G. J. Mendel, 1822– |
|
1884)). Менделевское наследование – |
|
наиболее объяснимый тип генетиче- |
|
ской передачи врождённых аномалий. |
|
При этом пороки могут передаваться |
|
либо по доминантному, либо по ре- |
|
цессивному типу наследования. |
|
Генотип каждого из родителей не- |
|
сёт два варианта (аллеля) гена, опре- |
|
деляющего данный признак, а ребёнок |
|
от каждого из родителей получает по |
|
одному аллелю. Проявление доми- |
|
нантного аномального признака воз- |
|
никает тогда, когда ребёнок наследу- |
|
ет от одного из родителей дефектный |
Рис. 7.1. Набор хромосом человека (кариотип |
ген, доминирующий над нормальным |
его определяется при наборе 23-й пары хро- |
вариантом от другого родителя. Ро- |
мосом: у мужчин – ХY, у женщин – ХХ) |
дитель с таким доминантным геном |
212
всегда имеет соответствующее нарушение (выраженное, возможно, в слабой форме). У ребёнка есть 50 %-ная вероятность получить данное нарушение в зависимости от того, нормальный или дефектный ген будет ему передан больным родителем. В качестве примеров доминантного наследования можно назвать болезнь Геттингтона (прогрессирующее поражение центральной нервной системы) или ахондропластическую карликовость (отставание роста костей).
Наследование рецессивного признака приводит к выраженному нарушению у ребёнка в том случае, когда оба родителя несут один и тот же дефектный ген (вместе с нормальным геном для данного признака), но клинического проявления заболевания у них нет. Каждый родившийся ребёнок будет иметь 25 %-ную вероятность не унаследовать дефектный ген ни от одного из родителей, 50 %-ную вероятность быть его носителем (обладая только одним дефектным геном) и 25 %-ную вероятность унаследовать его в «двойной дозе» (два дефектных гена), наследуя, таким образом, заболевание. Примеры рецессивного типа наследования – серповидноклеточная анемия (вызываемая дефектом в молекуле гемоглобина), талассемия (ещё одна форма анемии, встречающаяся в основном у лиц средиземноморского и азиатского происхождения), а также болезнь Тея–Сакса (нарушение обмена веществ, приводящее к смерти в раннем детском возрасте и проявляющееся в основном в семьях евреев, выходцев из Восточной Европы).
Нарушения, аналогичные рассмотренным выше, вызываются аутосомным геном (расположенным не в половых хромосомах), и потому их называют ау тосомными заболеваниями. К наиболее частым видам наследования признаков относят:
Аутосомнодоминантный тип наследования. Носитель патологического признака – доминантный ген, содержащийся в аутосоме (неполовой хромосоме). При этом типе наследования невозможно рождение больного ребёнка у здоровых родителей – хотя бы один из родителей страдает от того же заболевания. При этом мальчики и девочки подвержены заболеванию в равной степени. Такие дефекты развития, как правило, бывают негрубыми и после успешной коррекции не препятствуют нормальной жизни.
Аутосомнорецессивный тип наследования. Носитель патологического признака – рецессивный ген, содержащийся в аутосоме. При аутосомно-ре- цессивном механизме наследования ситуация выглядит парадоксально – у здоровых родителей рождается ребёнок с дефектами развития (порой тяжелейшими и даже несовместимыми с жизнью). Причина – носительство обоими супругами в скрытом состоянии мутантных рецессивных генов. При этом рождение больного ребёнка не обязательно означает, что все следующие дети будут страдать тем же заболеванием. Так же, как и при аутосомно-доминант- ном типе, мальчики и девочки подвержены заболеванию в равной степени.
Особым видом является сцепленное с полом рецессивное наследование.
Пороки развития, сцепленные с полом, обусловлены в основном рецессивны-
213
ми мутациями в женской половой хромосоме (этот тип наследования называют еще Х-хромосомным). Такой признак всегда передается через мать – носительницу рецессивного «больного» гена (сама женщина здорова). Практически все поражённые – мужчины (у поражённого гена Х-хромосомы в Y-хромосоме отсутствует «партнёр», который мог бы доминировать над ним). Больной мужчина никогда не передает заболевания своим сыновьям (ведь они получают от него «здоровую» Y-, а не мутантную Х-хромосому), однако все его дочери будут носительницами «рокового» гена. Цветовая слепота и гемофилия представляют собой X-сцепленные рецессивные нарушения. При другом X-сцепленном заболевании, называемом синдромом ломкой X-хромосомы, наблюдается различная степень умственной отсталости. Мужчины поражаются им чаще и в более тяжёлой форме.
Механизмы наследственно обусловленных аномалий. Причинными фак-
торами хромосомной патологии являются все виды хромосомных мутаций (хромосомные аберрации) и некоторые геномные мутации (изменения числа хромосом). При этом у человека встречаются только три типа геномных мутаций: тетраплоидия, триплоидия и анеуплоидия. Среди всех вариантов анеуплоидий наблюдаются только трисомии по аутосомам, полисомии по половым хромосомам (три-, тетра- и пентасомии), а из моносомий – только моносомия X.
В человеческой популяции наблюдаются все типы хромосомных мутаций: делеции (нехватка участка хромосомы), дупликации (удвоение участка), инверсии (поворот участка хромосомы на 180°) и транслокации (перенос части хромосомы с одной на другую).
Делеция в одной из гомологичных хромосом означает частичную моносомию по этому участку, а дупликация – частичную трисомию. Когда трансло кация является реципрокной (взаимной), без потери участков вовлеченных в неё хромосом, она называется сбалансированной. Данная мутация, как и ин версия, не проявляется у носителя фенотипически, так как при этом сохраня - ется баланс генов. Вместе с тем в процессе кросинговера у носителей сбалансированных транслокаций и инверсий могут образовываться несбалансированные гаметы, т. е. половые клетки с частичной дисомией, или с частичной нулисомией, или с обеими аномалиями в разных участках. В норме каждая гамета моносомна (имеет гаплоидный набор хромосом). При потере двумя акроцентрическими хромосомами коротких плеч и соединении их центромерами может образовываться одна метацентрическая хромосома. Эти транслокации называются робертсоновскими. При концевых делециях обоих плеч хромосомы (делеции теломеров) образуется кольцевая хромосома. У человека, унаследовавшего такие измененные хромосомы от одного из родителей, будет частичная моносомия по одному или двум концевым участкам хромосомы. В ряде случаев может происходить поперечный, а не продольный, как обычно, разрыв хроматид в области центромер. При этом формируются изохромосомы, представляющие собой зеркальное отображение двух одинаковых плеч
214
(длинных или коротких). Наличие у человека изохромосом проявляется фенотипически, так как имеют место одновременно и частичная моносомия (по отсутствующему плечу), и частичная трисомия (по имеющемуся плечу).
Хромосомные болезни встречаются у новорождённых детей с частотой примерно 2,4 случая на 1000 родившихся. Большинство хромосомных аномалий (полиплоидии, гаплоидии, трисомии по крупным хромосомам, моносомии) несовместимы с жизнью – эмбрионы и плоды элиминируются из организма матери в основном в ранние сроки беременности.
Хромосомные аномалии возникают и в соматических клетках с частотой около 2 %. В норме такие клетки устраняются (элиминируются) иммунной системой, если они проявляют себя чужеродно. Однако в некоторых случаях (при активации онкогенов) хромосомные аномалии могут быть причиной злокачественного роста. Так, транслокация между 9-й и 22-й хромосомами вызывает миелолейкоз.
Патогенез хромосомных болезней ещё до конца не выясен. Специфические эффекты связаны с изменением числа структурных генов, кодирующих синтез специфических белков (увеличение при трисомиях и уменьшение при моносомиях). Полуспецифические эффекты при хромосомных болезнях могут быть обусловлены изменением числа генов, которые и в норме представлены многочисленными копиями (гены тРНК, рРНК, гистоновых и рибосомных белков и т. п.). Неспецифические эффекты хромосомных аномалий связывают с содержанием гетерохроматина, играющего важную роль в делении клеток, их росте и других физиологических процессах.
Общей закономерностью всех форм хромосомных болезней является множественность поражения. В первую очередь это черепно-лицевые поражения, врождённые пороки развития систем органов, замедленные внутриутробные и постнатальные рост и развитие, отставание в психическом развитии, нарушение функций нервной, иммунной и эндокринной систем. Фенотипические проявления хромосомных мутаций зависят от следующих главных факторов:
1)характеристики вовлечённой в аномалию хромосомы (специфический набор генов);
2)типа аномалии (трисомия, моносомия, полная, частичная);
3)размера недостающего (при частичной моносомии) или избыточного (при частичной трисомии) генетического материала;
4)степени мозаичности организма по аберрантным клеткам;
5)генотипа организма;
6)условий внешней среды.
В настоящее время установлено, что при хромосомных мутациях наиболее специфичные для того или иного синдрома проявления обусловлены изменениями небольших участков хромосом. Например, специфические симптомы болезни Дауна обнаруживаются при трисомии небольшого сегмента длинного плеча 21-й хромосомы (21q22.1), синдрома «кошачьего крика» – при делеции средней части короткого плеча 5-й хромосомы (5р15), синдрома Эд-
215
вардса – при трисомии сегмента длинного плеча хромосомы. Окончательный диагноз хромосомных болезней устанавливается цитогенетическими методами.
Трисомии. Наиболее часто у человека встречаются трисомии по 21, 18 и 13-й паре хромосом (эпонимы – болезнь Дауна, синдромы Эдвардса и Патау соответственно) (рис. 7.2).
Таблица 7.2. Характеристика наиболее частых трисомий
Изучаемый показатель |
Трисомия 21 |
Трисомия 18 |
Трисомия 13 |
|
|
|
|
|
|
Частота |
1:800 |
1:8000 |
1:15 000 |
|
возникновения |
||||
|
|
|
||
Тонус мышц |
Гипотония |
Гипертония |
Гипоили гипертония |
|
Череп/головной мозг |
Умеренная микроцефа- |
Микроцефалия, вы- |
Микроцефалия, ско- |
|
|
лия, плоский затылок, |
ступающий затылок |
шенный затылок, де- |
|
|
три родничка |
|
фекты кожи в области |
|
|
|
|
свода черепа и в об- |
|
|
|
|
ласти затылка |
|
Глаза |
Раскосые глаза, складки |
Узкая глазная щель, |
Микрофтальмия, ги- |
|
|
эпиканта, пятнистая ра- |
помутнение роговицы |
потелоризм, колобома |
|
|
дужная оболочка (пятна |
|
радужной оболочки, |
|
|
Брашфильда) |
|
дисплазия сетчатки |
|
Уши |
Низко посажены, допол- |
Низко посажены, по- |
Низко посажены, по- |
|
|
нительные складки на |
роки развития |
роки развития |
|
|
верхнем завитке |
|
|
|
Лицо |
Выпадающий язык, |
Маленький рот, ми- |
Расщепление губы |
|
|
большие щёки, плоское |
крогнатия |
и нёба |
|
|
переносье |
|
|
|
Скелет |
Клинодактилия мизин- |
Сжатие кистей рук в |
Заднеаксиальная по- |
|
|
ца, большое расстояние |
кулак, отсутствие дис- |
лидактилия, плоские |
|
|
между первым и вторым |
тальной складки на |
ногти, сжатие кистей |
|
|
пальцем ноги, избыточ- |
мизинце, гипоплазия |
рук в кулак |
|
|
ное количество кожи на |
ногтей, малый рост, |
|
|
|
задней поверхности шеи, |
тонкие рёбра |
|
|
|
малый рост |
|
|
|
Пороки сердца |
40 % |
60 % |
80 % |
|
Выживаемость |
Высокая |
90 % погибают на пер- |
80 % погибают на |
|
|
|
вом году жизни |
первом году жизни |
|
Наличие других |
|
Изогнутая стопа, по- |
Пороки развития по- |
|
признаков |
|
ликистоз почек, дер- |
ловых органов, поли- |
|
|
|
матоглифика – дуги |
кистоз почек, увели- |
|
|
|
|
чение выступов на |
|
|
|
|
ядрах нейтрофилов |
Аномалии сочетания половых хромосом. Пол будущего ребёнка определя-
ется в момент оплодотворения и зависит от сочетания половых хромосом (XX – женский организм, XY – мужской). При нарушении течения митоза могут образовываться необычные особи – гинандроморфы. Содержание половых хромосом в разных клетках таких особей может быть разное (мозаицизм). У человека могут быть разные случаи мозаицизма: ХХ/ХХХ, XY/XXY,
216
Х0/ХХХ, X0/XXY и др. Степень клинического проявления зависит от количества мозаичных клеток – чем их больше, тем сильнее проявление. При нормальном течении мейоза в женском организме образуется один тип гамет, содержащих Х-хромосому. Однако при нерасхождении половых хромосом могут образовываться еще два типа гамет – XX и 0 (не содержащая половых хромосом). У мужского организма в норме образуется два типа гамет, содержащих Х- и Y-хромосомы. При нерасхождении половых хромосом возможны варианты гамет – XY и 0.
Вероятные комбинации половых хромосом в зиготе у человека:
1.XX – нормальный женский организм.
2.XXX – синдром трисомии X. Частота встречаемости 1:1000. Кариотип 47, ХХХ. В настоящее время имеются описания тетра- и пентосомий X. Трисомия по Х-хромосоме возникает в результате нерасхождения половых хромосом в мейозе или при первом делении зиготы.
3.Синдрому полисемии X присущ значительный полиморфизм – женский организм и мужеподобное телосложение. Могут быть недоразвиты первичные и вторичные половые признаки. В 75 % случаев у пациентов наблюдается умственная отсталость умеренной степени. У некоторых из них нарушена функция яичников (вторичная аменорея, дисменорея, ранняя менопауза). Иногда такие женщины могут иметь детей. Повышен риск заболевания шизофренией. С увеличением числа дополнительных Х-хромосом нарастает степень отклонения от нормы.
4.Х0 – синдром Шерешевского–Тернера (моносомия X).
5.XY – нормальный мужской организм.
6.XXY и XXXY – синдром Клайнфелтера.
7.Y0 и 00 – зиготы нежизнеспособны.
8.Вероятны случаи увеличения количества Y-хромосом: XYY, XXYY и др. При этом пациенты имеют признаки синдрома Клайнфелтера, высокий рост (в среднем 186 см), отличаются агрессивным поведением. Могут быть аномалии зубов и костной системы. Половые железы развиты нормально. Чем больше в наборе Y-хромосом, тем значительнее снижение интеллекта.
Синдромы частичных анеуплодий. Кроме полных трисомий и моносомий известны синдромы, связанные с частичными трисомиями и моносомиями практически по любой хромосоме. Однако частота встречаемости этих синдромов реже 1 случая на 100 000 рождений.
Синдром трисомий по короткому плечу 9-й хромосомы (9р+) – наиболее частая форма частичных трисомий (описано свыше 200 случаев). Для пациентов с трисомией 9р+ характерны умственная отсталость, задержка роста, микроцефалия, антимонголоидный разрез глазных щелей, глубоко посаженные глаза, опущенные уголки рта, нос с характерным округлым кончиком, низко расположенные оттопыренные ушные раковины, недоразвитие ногтей и дистальных фаланг пальцев рук. Часто наблюдаются выступающие лобные кости, повышенное оволосение, пятна цвета кофе с молоком на коже, эпикант,
217
косоглазие, высокое дутообразное нёбо, короткая шея, сколиоз, частичная синдактилия пальцев стоп. Примерно в четверти случаев обнаруживаются врождённые пороки сердца. Прогноз для жизни сравнительно благоприятный (описаны случаи, когда пациенты достигали преклонного возраста).
Распространённость синдромов частичных моносомий примерно такая же, как и синдромов частичных трисомий. Наиболее известные из них – синдромы Вольфа–Хиршхорна, «кошачьего крика», Орбели и Прадери–Вилли. При этом синдром «кошачьего крика» (5р–) обусловлен делецией короткого плеча 5-й хромосомы. Популяционная частота синдрома – примерно 1:45 000. Для данного синдрома наиболее характерны специфический плач, напоминающий кошачье мяуканье, лунообразное лицо, мышечная гипотония, умственное и физическое недоразвитие, микроцефалия, низко расположенные, иногда деформированные ушные раковины, эпикант, антимонголоидный разрез глазных щелей и косоглазие. Иногда наблюдаются атрофия зрительного нерва и очаги депигментации сетчатки. Как правило, выявляются пороки сердца. Наиболее постоянный признак синдрома – «кошачий крик» (обусловлен изменениями гортани – сужением, мягкостью хрящей, отёчностью или необычной складчатостью слизистой оболочки, уменьшением надгортанника). Изменения других органов и систем неспецифичны. Продолжительность жизни у пациентов с этим синдромом значительно снижена, только около 14 % из них переживают возраст 10 лет. О других синдромах речь пойдёт ниже.
2. Внешние воздействия. Лекарства. После выявления в середине 1960-х годов причины развития врождённых аномалий, обусловленных приёмом беременными лекарственного препарата талидомид, стало ясно – многие лекарства могут преодолевать плацентарный барьер и воздействовать на эмбрион или плод. Особенно опасным сроком для возникновения пороков является ранний эмбриональный период, когда формируется большинство структур организма. Хотя основные физические аномалии возникают начиная со 2-й
идо 8-й недели беременности, отдельные аномалии глаз, внутреннего уха
инервной системы могут проявиться и позже (уже не у эмбриона, а у плода). До 2-й недели воздействие вредных веществ блокирует имплантацию эмбриона в маточную стенку либо столь сильно влияет на него, что дальнейшее его развитие невозможно.
Лекарства с доказанным тератогенным эффектом (во время беременности они абсолютно противопоказаны) и препараты, которые могут нанести вред плоду:
аминоптерин (вызывает множественные аномалии, постнатальную задержку развития плода, аномалии лицевого отдела черепа, смерть плода);
андрогены (приводят к вирилизации, укорочению конечностей, аномалиям трахеи и пищевода, дефектам сердечно-сосудистой системы);
диэтилстилбестрол (вызывает развитие аденокарциномы влагалища, дефекты шейки матки, пениса, гипотрофию яичек);
стрептомицин (способствует развитию глухоты);
218
дисульфирам (приводит к спонтанным абортам, расщеплению конечностей, косолапости);
эрготамин (вызывает спонтанные аборты, симптомы раздражения ЦНС); эстрогены (способствуют развитию врождённых пороков сердца, феми-
низации мужского плода, возникновению аномалий сосудов); галотан, фторотан (приводят к спонтанным абортам); йод131 (способствует развитию кретинизма, гипотиреоза);
метилтестостерон (вызывает маскулинизацию женского плода); прогестины (вызывают маскулинизацию женского плода, увеличение кли-
тора, способствуют развитию пояснично-крестцовых сращений); хинин (обладает ототоксическим эффектом, приводит к задержке психиче-
ского развития, врождённой глаукоме, развитию аномалий мочеполовой системы и к смерти плода);
талидомид (вызывает пороки конечностей, аномалии сердца, почек и же- лудочно-кишечного тракта);
триметадион (при использовании во время беременности приводит к аномалиям сердца, глаз, задержке психического развития; после его применения у новорождённого отмечается характерное лицо – V-образные брови и низко поставленные глаза);
ретиноиды (изотретиноин, роанккутан, этретинат, тигазон, ацитретин) способствуют возникновению аномалий конечностей, лицевого отдела черепа, сердца и ЦНС, мочеполовой системы, недоразвитию ушных раковин);
тетрациклин (ведёт к дисколорации зубов, гипоплазии зубной эмали ребёнка);
литий (способствует развитию врождённых заболеваний сердца, зоба, гипотонии, цианоза);
диазепам (приводит к гипотермии, гипотонии, раздвоению и аномалиям конечностей);
имипрамин (способствует развитию нарушений со стороны органов дыхания, приводит к дефектам конечностей, тахикардии, задержке мочи, развитию неонатального дистресс-синдрома);
нортриптилин (вызывает развитие неонатального дистресс-синдрома, цианоз, тремор, задержку мочи);
аспирин (приводит к неонатальным кровотечениям, внутричерепным кровотечениям у недоношенных, стойкой гипертензии лёгочной артерии);
индометацин (вызывает гипертензию лёгочных артерий, нарушение сер- дечно-лёгочной адаптации, смерть плода);
варфарин (приводит к эмбриопатии, задержке развития, атрофии зритель - ного нерва, судорогам, кровотечению, нередко заканчивающемуся летальным исходом).
фенобарбитал (способствует ухудшению слуха, угнетению ЦНС, анемии, тремору, синдрому отмены, артериальной гипертензии);
219

фенитоин (приводит к аномалиям конечностей и черепно-лицевого отдела позвоночника, задержке умственного развития, врождённым заболеваниям сердца, кровотечениям); вальпроат натрия (вызывает расщелины
позвоночника); этосуксимид (приводит к монголоидной
внешности, короткой шее, лишним соскам (рис. 7.2), задержке развития, дермоидной фистуле);
хлоротиазид (способствует развитию холестаза, панкреатита);
резерпин (обладает ототоксическим действием на плод);
азатиоприн (вызывает стеноз лёгких, по - лидактилию, приводит к лицевому дисморфогенезу);
бусульфан (вызывает задержку внутриутробного и послеродового развития, помутнение роговицы);
|
хлорамбуцил (приводит к нарушениям функ- |
|
Рис. 7.2. Многососковость, наблю- |
ции почек); |
|
5фторурацил (вызывает спонтанные абор- |
||
даемая после приёма беременной |
||
ты, дефекты лицевого отдела черепа); |
||
мамой этосуксимида (аномальные |
||
соски располагаются по эмбриональ- |
колхицин (способствует выкидышам, воз- |
|
ным линиям Гешикташа) |
никновению трисомии 21); |
|
Приведено по: Н. Б. Ситковский |
меркаптопурин (приводит к спонтанным |
|
с соавт. (1981) |
абортам, дефектам лицевого отдела черепа); |
|
|
метотрексат (вызывает отсутствие лоб - |
ной кости, сращение костей черепа, спонтанные аборты, задержку послеродового развития);
винкристин (вызывает внутриутробное недоразвитие плода, неправильное его положение в матке);
метимизол (приводит к развитию зоба, изъязвлениям срединного отдела волосистой части головы);
хлорпропамид (способствует развитию множественных пороков развития, гипогликемии);
хлордиазепоксид (приводит к депрессии, изменению сознания, синдрому отмены, гипервозбудимости);
мепробамат (вызывает врождённые дефекты сердца, синдром отмены, пороки диафрагмы);
витамин А (в дозах свыше 10 000 ME/сут приводит к развитию дефектов сердечно-сосудистой системы, ушных раковин и др.).
220