

Ч 2
.pdfОписана французскими врачами G. Hayem (1841–1933) и G. F. I. Widal (1862–1929).
Болезнь Гербази (синонимы – анемия Гербази, синдром Гербази). Бо-
лезнь неясной этиологии, развивающаяся у детей грудного возраста. По клиническим проявлениям напоминает пернициозную анемию, но, в отличие от последней, не связана с недостаточностью гастромукопротеина и через некоторое время самопроизвольно излечивается.
Описана итальянским педиатром М. Gerbasi.
Болезнь (синдром) Дресслера I (синоним – пароксизмальная гемогло-
бинурия). После переохлаждения или физического напряжения отмечаются озноб, лихорадка, головная боль, боль в пояснице, желтуха. Может быть умеренное увеличение печени и селезёнки. Моча тёмно-бурого цвета из-за большого количества метгемоглобина. В крови анемия, гипербилирубинемия, эозинофилия и лимфоцитоз. Болезнь протекает приступами. Предполагается аутоиммунный механизм её развития.
Описана американским врачом W. Dressler.
Болезнь (синдром) Мошковича (синонимы – тромбоцитопенический акроангиотромбоз, тромботическая микроангиопатия, синдром Мошко- вича–Зингера–Симмерса, синдром Мошковича–Зингера, тромботическая пурпура, тромбоцитопеническая тромбогемолитическая пурпура). Острая болезнь, вероятно, аутоиммунной природы, характеризующаяся сочетанием гемолитической анемии с тромбоцитопенией и геморрагическим синдромом.
Описана американским врачом Е. Moschcowitz (1879–1964).
Болезнь Уилкинсона (синоним – болезнь Израэльса–Уилкинсона). B12-
фолиево-ахрестическая анемия, возникающая вследствие нарушения процесса ассимиляции цианокобаламина (витамина B12) и фолиевой кислоты костным мозгом.
Описана в 1926 г. английским гематологом J. F. Wilkinson.
Болезнь Херика–Полинга (синонимы – серповидноклеточная анемия, гемоглобиноз S, «реологическая болезнь»). Заболевание относится к гемо-
глобинопатиям. Широко распространено в странах Средиземноморья, Ближнего и Среднего Востока, Индии, Америке (рис. 3.10). Описано после открытия эритроцитов серповидной формы и получения доказательств, что НbS отличается от НbА тем, что в положении 6 в цепи глютаминовая кислота заменена на валин.
Заболевание наследуется по аутосомному типу и носит кодоминантный характер. Поскольку нарушения, обусловленные гомозиготностью, выявляются у обоих родителей, показатели заболеваемости и смертности очень высоки. Замещение глютамина валином приводит к тому, что у НbS вместо отрицательного заряда, характерного для НbА, появляется нейтральный, что усиливает связь одной молекулы гемоглобина с другой. Внутри эритроцита гемоглобин переходит в состояние геля, а при пониженном парциальном давлении кислорода осаждается в виде тактоидов – веретенообразных остроко-
111
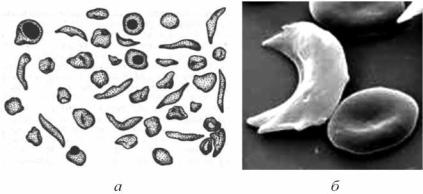
Рис. 3.10. Внешний вид эритроцитов (а) и их электронная микрофотограмма (б) при болезни Херика–Полинга
Приведено по: Г. Е. Ройтберг, А. В. Струтынский (1999)
нечных кристаллов, которые растягивают эритроциты, придавая им серповидную форму. Появление серповидных эритроцитов значительно повышает вязкость крови, что уменьшает скорость кровотока и приводит к закупорке мелких капилляров. S-эритроциты теряют пластичность, застревают в капиллярах, вызывая тромбоз (окклюзию) сосудов. Возникают инфаркты, сопровождаемые гипоксией. У пациентов выявляются бледность кожи и слизистых оболочек, желтушность, усиливающаяся с возрастом.
У детей младшего возраста, начиная с 6 мес., пальпируется селезёнка, однако к 8 годам спленомегалию обнаруживают редко (это объясняется фиброзом селезёнки, развивающимся на фоне частых инфарктов). У 60 % детей выявляется гепатомегалия, нередко – кардиомегалия (при этом над областью сердца выслушивают различной интенсивности шумы). Характерным симптомом является аденопатия. В желчном пузыре определяются камни. Часто встречается язва двенадцатиперстной кишки. Пациенты имеют характерный вид – удлинённый нижний сегмент тела, дорсальный кифоз и люмбальный лордоз, куполообразное («готическое») нёбо, выступающий лоб и башнеобразный череп, значительное удлинение конечностей. В возрасте до 3 лет, как правило, нет отставания в росте от сверстников, затем имеет место некоторая задержка до 7–8 лет, а во взрослом возрасте рост у пациентов нормальный и даже несколько выше. Дети отстают в половом развитии, однако уровень интеллекта, как правило, нормальный.
Заболевание протекает хронически, пациенты с тяжёлой формой живут около 20 лет. Бывают периодически острые состояния (кризы), которые де - лятся на две группы: 1) клинические (болевые или вазоокклюзионные), при которых показатели состава гемоглобина и ретикулоцитов не отличаются от нормы; 2) гематологические с резким снижением уровня гемогло - бина и высоким ретикулоцитозом. Нередко кризы сочетаются. Пренатальная диагностика позволяет установить диагноз у плода с 10–12 недель (при
112
биопсии трофобласта). С 16–20 недель |
|
|||
возможно проведение аспирации из |
|
|||
вен плаценты. |
|
|
||
Серповидноклеточная |
анемия |
|
||
впервые была описана в 1910 г. амери- |
|
|||
канским врачом J. B. Herik. Наличие |
|
|||
патологического гемоглобина S, со- |
|
|||
держащего в одном из участков бел- |
|
|||
ковой цепи аминокислоту валин вме- |
|
|||
сто глутаминовой кислоты, лежащей |
|
|||
в основе заболевания, было установ- |
|
|||
лено в 1949 г. американским учёным |
|
|||
L. Poling, что положило начало уче- |
|
|||
нию о молекулярных болезнях чело- |
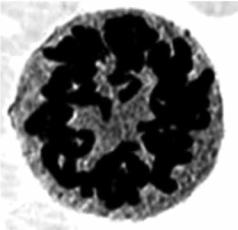
Рис. 3.11. Делящийся эритробласт (избыточ- |
|||
века. |
|
|
||
Болезнь Эклина. Доброкачествен- |
ное скопление этих клеток в периферической |
|||
крови отмечается при болезни Эклина) |
||||
ная анемия новорождённых, характе- |
||||
Приведено по: Г. Е. Ройтберг, А. В. Струтын- |
||||
ризующаяся |
гепатоспленомегалией |
ский (1999) |
||
и желтухой, |
а иногда – появлением |
|
||
небольшого количества эритробластов (рис. 3.11) в периферической крови. Описана швейцарским педиатром Th. Ecklin.
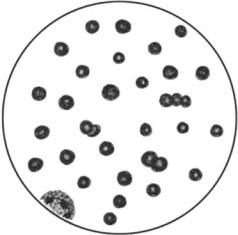
Кольца Кебота. Морфологические образования в эритроцитах в форме кольца, восьмерки или скрипичного ключа, являющиеся, вероятно, остатками ядерной оболочки. Встречаются при некоторых анемиях.
Описаны американским врачом R. С. Cabot (1868–1939).
Критерии дефицита железа по Демидову. Критерии оценки анемии в за-
висимости от дефицита сывороточного железа и ферритина представлены в табл. 3.2.
Таблица. 3.2. Критерии оценки анемии в зависисмости
от дефицита сывороточного железа и ферритина
Показатель |
Норма |
Латентный |
Железодефицитная |
|
|
|
дефицит железа |
анемия |
|
|
|
|
|
|
Гемоглобин, г/л: |
|
|
|
|
мужчины |
130–160 |
> 130 |
< 130 |
|
женщины |
120–150 |
> 120 |
< 120 |
|
|
||||
Сывороточное железо, мкмоль/л: |
|
|
|
|
мужчины |
10,6–28,3 |
< 7,5 |
< 7,5 |
|
женщины |
6,6–26,0 |
< 6,0 |
< 6,0 |
|
|
||||
Ферритин, нг/мл: |
|
|
|
|
мужчины |
34–310 |
< 40 |
< 12 |
|
женщины |
||||
22–112 |
< 40 |
< 12 |
||
|
Предложены в 1993 г. врачом А. В. Демидовым.
113
Синдром Бернара. Разновидность спонтанной энзимопенической гемолитической желтухи, анемии с нарушением белкового состава плазмы крови. Заболевание начинается с недомогания, лихорадки, артралгии. Затем присоединяются боли в животе, тошнота, рвота, желтуха. Характерны выраженная анемия и гемоглобинурия, аномалий со стороны эритроцитов не отмечается. Зарегистрированы случаи семейной формы заболевания.
Описан в 1946 г. французским гематологом J. Bernard.
Синдром (болезнь) Гассера (синонимы – болезнь Гассера–Каррера, ге-
молитико-уремический синдром). Болезнь почек, протекающая с гемолитической анемией, тромбоцитопенией и острой почечной недостаточностью.
Описан в 1956 г. швейцарским педиатром С. Gasser.
Синдром Генсслена (синоним – семейный гемолитический желтушно-
костный синдром). Наследственное заболевание, характеризующееся выраженными метаболическими изменениями в костях и гемолитической анемией. Проявляется в любом возрасте. Гемолитическую анемию сопровождает спленомегалия. Отмечаются изменения в крупных костях, характерными являются уменьшение или отсутствие перекладин и значительное утолщение кортикального слоя с остеопорозом длинных трубчатых костей. Кортикальные изменения развиваются из-за возвратной и хронической гиперплазии кроветворных костномозговых тканей. Могут наблюдаться остеопороз и гиперостоз черепа, башнеобразный череп, неправильное положение зубов, брахиодакти-
лия, полисиндактилия и врождённый вывих бедра.
Описан американским хирургом
|
F. J. Gaenslen (1877–1937). |
|
|
Синдром (болезнь) Ледерера– |
|
|
Брилла (синоним – острая гемолити- |
|
|
ческая анемия). После нескольких |
|
|
дней продромального периода (катар |
|
|
верхних дыхательных путей, головная |
|
|
боль, понос) появляются сильный оз- |
|
|
ноб, высокая температура, рвота и гема- |
|
|
турия. Быстро прогрессирует желтуха. |
|
|
В некоторых случаях присоединяются |
|
|
приступообразная боль в животе, уме- |
|
|
ренное увеличение селезёнки, печени |
|
|
и лимфоузлов, симптомы геморрагиче- |
|
|
ского диатеза. В крови малокровие |
|
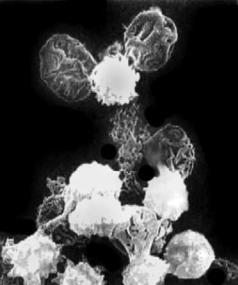
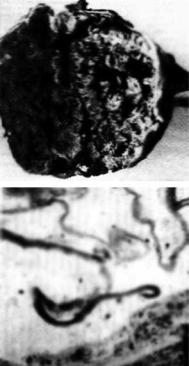
Рис. 3.12. Острый гемолиз крови при синдро- |
с мегалоцитозом, ритикулоцитозом, |
|
ме (болезни) Ледерера–Брилла (данные элек- |
значительный лейкоцитоз со сдвигом |
|
тронной микроскопии) |
влево до миелобластов. Гемолиз при этом |
|
Приведено по: http://student.km.ru/ref_show_ |
||
заболевании внеклеточный (рис. 3.12). |
||
frame.asp?id= 1BC80D01E0464E578EE31F0E |
Этиология не известна. |
|
A59ECB26 |
114
|
шены. В течении заболевания выделя- |
|
|
ют: 1) гемолитический криз (проявля- |
|
|
ется нарастанием анемии с одышкой, |
|
|
тошнотой, рвотой, болями в животе, |
|
|
адинамией, повышением температу- |
|
|
ры тела до 38–40 °С, увеличением пе- |
|
|
чени и селезёнки), при котором в кро- |
|
|
ви отмечается нормохромная анемия |
|
|
(ретикулоциты до 50–60 % и выше); |
|
|
2) апластический криз (характерны |
|
|
симптомы тяжёлой анемии без при- |
|
|
знаков желтухи, небольшие размеры |
|
|
селезёнки, гипохромная анемия, отсут- |
|
|
ствие ретикулярной реакции). В ряде |
|
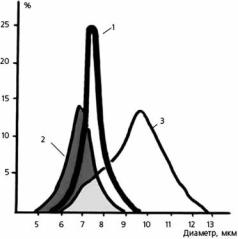
Рис. 3.14. Кривая Прайса–Джонса (количе- |
случаев при данном синдроме отме- |
|
ственное соотношение эритроцитов разно- |
чают башнеобразный или большой |
|
го диаметра): 1 – у здорового человека; 2 – |
круглый череп, малый рост, аномалии |
|
у пациента с железодефицитной анемией; 3 – |
развития зубов, глаз и ушей, врождён- |
|
у пациента с В12-фолиеводефицитной анемией |
ные пороки сердца, высокое нёбо, за- |
|
Приведено по: Г. Е. Ройтберг, А. В. Струтын- |
||
падение переносицы, кривошею, поли- |
||
ский (1999) |
и синдактилию. |
|
|
Диагноз наследственного микросфероцитоза ставят на основании течения заболевания (чередование кризов и ремиссий), клинической картины (желтуха, спленомегалия, боли в правом подреберье, анемия), данных исследования периферической крови пациентов (нормохромная анемия, ретикулоцитоз, микросфероцитоз). Важное значение имеет обследование их родственников, у которых могут определяться едва уловимые признаки гемолиза или микросфероцитоз без клинических проявлений. Дополнительными диагностическими критериями могут служить некоторые лабораторные тесты. Характерным лабораторным признаком заболевания является снижение осмотической резистентности эритроцитов по отношению к гипотоническим растворам хлористого натрия. Начало гемолиза при наследственном микросфероцитозе соответствует 0,6–0,7 %, конец – 0,4 % (в норме 0,48 и 0,22 % соответственно). Снижение осмотической резистентности свидетельствует о преобладании в крови эритроцитов сферической формы – сфероцитов, которые менее стойки к осмотическому гемолизу, чем нормальные макропланоциты. Эритроциты больных наследственным микросфероцитозом легко разрушаются после суточной инкубации дефибринированной крови в термостате при 37 °С. Добавление к эритроцитам глюкозы значительно уменьшает гемолиз, в то время как АТФ не влияет на него.
Дифференциальная диагностика наследственного микросфероцитоза сводится прежде всего к диагностике гемолитических анемий вообще и требует исключения целого ряда заболеваний (аутоиммунной гемолитической ане-
116
мии, наследственного микросфероцитоза, дефицита глюкозо-6-фосфатдегид- рогеназы, болезни Маркиафавы–Микели, талассемии).
Описан в 1900 г. польским врачом О. Minkowski (1858–1931), французским врачом А. М. Е. Chauffard (1855–1932) и американским хирургом F. J. Gaenslen (1877–1937).
Синдром (болезнь) Ниманна–Пика. Заболевание с аутосомно-рецессив- ным типом наследования и неуклонно прогрессирующим течением. Наследственный липоидоз, обусловленный нарушением обмена сложных липидов с накоплением сфингомиелина в клетках ретикулоэндотелиальной и центральной нервной систем, который придаёт последним характерный пенистый вид (клетки Пика). Заболевание проявляется сразу после рождения. Дети становятся малоподвижными, вялыми, апатичными, отказываются от груди. Периодически появляется рвота, отмечается отставание в физическом и психическом развитии, может наблюдаться умеренная гидроцефалия. Периоды апатии сменяются беспокойством, гипертермическими кризами и приступами асфиксии. Наблюдаются прогрессивное похудание ребёнка, связанное с потерей аппетита, увеличение размеров живота, гепато- и спленомегалия, асцит.
Клинически различают три типа болезни: тип А (более 1/2 всех наблюдений) – проявляется с первых месяцев жизни выраженной гепато- и спленомегалией, задержкой физического и умственного развития, тяжёлыми неврологическими расстройствами, большинство детей погибает в возрасте до 3 лет; тип В (висцеральная, или хроническая, форма заболевания) – отличается поздним началом и распространённым поражением внутренних органов, при этом поражения нервной системы, как правило, не наблюдается; тип С (подострая, или юношеская, форма заболевания) – характеризуется медленным прогрессированием неврологической симптоматики, гепато- и спленомегалией, анемией, судорогами, мозжечковыми расстройствами. Прогноз заболевания неблагоприятный.
Описан в 1914 г. немецким педиатром А. Niemann (1880–1921), а затем в 1922 г. – немецким патологоанатомом L. Pieck (1868–1944).
Синдром Пламмера–Винсона (синонимы – синдром Патерсона–Келли,
сидеропенический синдром). Предполагается, что болезнь обусловлена недостаточностью рибофлавина и фолиевой кислоты. Характеризуется дисфагией, атрофией слизистой оболочки пищеварительного тракта, дистрофией ногтей, гипохромной анемией, уменьшением содержания железа в крови.
Описан в 1932 г. американским врачами Н. S. Plummer (1874–1937)
и P. P. Vinson.
Синдром (болезнь) Странского–Регалы. Общее название различных форм семейной гемолитической анемии, наблюдаемых у жителей Малайских островов.
Описан в 1939 г. американскими педиатрами Е. Stransky и А. С. Regala.
Синдром Фабера (синоним – синдром Гайема–Фабера). Эссенциальная железодефицитная анемия с ахлоргидрией, проявляющаяся дисфагией и глос-
117
ситом, общей слабостью, малокровием, появлением болезненных трещин в углах рта и на пальцах, остеопорозом и задержкой окостенения скелета.
Описан французским врачом G. Hayem (1841–1933) и датским врачом К. Faber (1862–1956).
Синдром Фелти. Сочетание полиартрита с увеличением селезёнки и лимфатических узлов, лейкопенией, иногда с анемией и тромбоцитопенией. Представляет собой форму ревматоидного артрита у взрослых (встречается преимущественно у женщин).
Описан в 1927 г. американским врачом A. R. Felty.
Синдром Циве. Проявляется гиперлипидемией, гемолитической анемией, желтухой и жировым гепатозом. Заболевание возникает у злоупотребляющих алкоголем лиц обоего пола моложе 50 лет. Развивается постепенно – нарастают астения, анорексия, похудание, диспепсические расстройства (тошнота, рвота, понос), боли в правом подреберье и подложечной области, гипертермия. Желтуха носит гемолитический характер, но может быть связана с повреждением паренхимы печени и холестазом. Гиперлипидемия сопровождается увеличением в сыворотке крови концентрации общих липидов, жирных кислот, триглицеридов и фосфолипидов (механизм гиперлипидемии обусловлен как стеатозом печени, так и алкогольным поражением поджелудочной железы). Усиленный гемолиз носит экстракорпускулярный характер и обусловлен гиперлипидемией. Жировая дистрофия печени отличается многообразием (нередко имеет место воспалительный синдром). Часто наблюдают телеангиэктазии (иногда паукообразные).
Описан в 1958 г. врачом L. Zieve.
Синдром (болезнь) Цюльцера–Огдена. Гипохромная анемия у детей грудного возраста, обусловленная недостатком в питании цианокобаламина, фолиевой и аскорбиновой кислот. Характеризуется геморрагическим диатезом, мегалобластической формой гемопоэза, гепатоспленомегалией.
Описан в 1964 г. американскими педиатрами W. W. Zuelzer и F. N. Ogden. Синдром Чедиака–Хигаши. Редкое наследственное заболевание обмена веществ, наследуемое по аутосомно-рецессивному типу. Проявляется аномалиями лейкоцитов и расстройствами пигментного обмена (гиперпигментацией и пигментной дистрофией). Кожа светлая, прозрачная; волосы светлые, сухие и редкие. Иногда выявляется гиперпигментация участков кожи после инсоляции. Живот увеличен в размерах. Отмечаются гепатоспленомегалия, анемия, лейкопения, тромбоцитопения, аномальная зернистость лейкоцитов
и лимфоцитов.
Прогноз неблагоприятный – если пациенты доживают до 20 лет, развивается лимфопролиферативное состояние, характеризующееся множественными инфильтратами во внутренних органах.
Описан в 1952–1954 гг.
118
3.2.Заболевания селезёнки и их проявления
Вклиническом плане различают следующие хирургические заболевания селезёнки:
1. Врождённые аномалии (гипоплазия; агенезия; добавочная селезёнка, многодольчатая селезёнка; врождённые обменные аномалии и ферментопатии, протекающие с явлениями спленомегалии и гиперспленизма, – болезнь Бюргера–Грютца, болезнь Гоше, болезнь Гюнтера, синдром Гельмгольца– Харрингтона и др.).
2. Вторичные поражения селезёнки на почве приобретённых заболеваний, протекающие с явлениями спленомегалии и гиперспленизма (изменения со стороны селезёнки при циррозе печени, синдроме Банти, гематологических синдромах, других поражениях гепатопанкреатодуоденальной зоны).
3. Травмы селезёнки (закрытые, открытые).
4. Опухоли селезёнки (первичные, вторичные; доброкачественные, злокачественные).
5. Паразитарные заболевания селезёнки (рис. 3.15).
6. Инфекционные заболевания селезёнки (гнойный спленит, абсцесс селезёнки).
7. Приобретённые кисты селезёнки – посттравматические (рис. 3.16), поствоспалительные, паразитарные; истинные, ложные; неосложнённые и осложнённые воспалением, нагноением, разрывом, кровотечением, сдавлением соседних органов и т. д.
Болезнь Бюргера–Грютца. Врождённое
(семейное) заболевание, в основе которого лежит нарушение липидного обмена. Основные проявления – гепатоспленомегалия, панкреатит, гиперлипидемия, ксантоматозные высыпания на коже в области локтевых и коленных суставов. В крови повышены уровни свободного холестерола и лецитина.
Описана немецкими врачами М. Burger (1885–1966) и О. Griitz (1886–1963).
Болезнь Гоше (синонимы – спленомегалия Гоше, керазиновая болезнь). Врождён-
ное (нередко семейное) заболевание. Этиология не известна. В основе заболевания лежит врождённый дефицит ферментов, обеспечи-
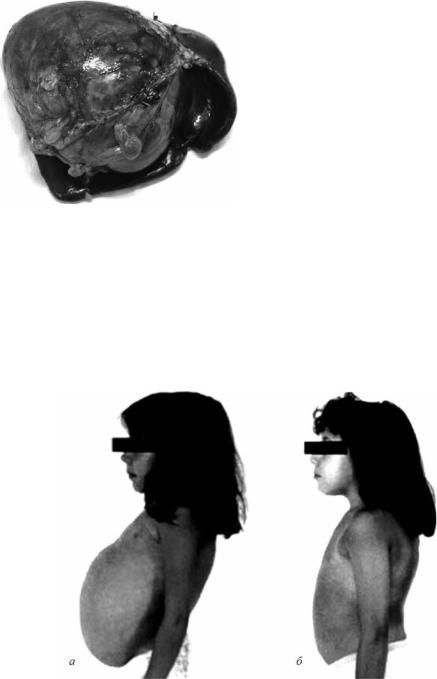
Рис. 3.15. Альвеококкоз селезёнки (а – макропрепарат удалённого органа; б – гистологические изменения)
Приведено по: http://www.surgerycom.net/Galery/Photo.htm
119
|
вающих отщепление глюкозы от глю- |
|
коцереброзидов с накоплением по- |
|
следних в клетках ретикуло-эндоте- |
|
лиальной системы (рис. 3.17). При |
|
этом нарушается белковый и липид- |
|
ный обмен, появляются гигантские |
|
клетки Гоше со скоплением керозина |
|
(глюкоцереброзида) в цитоплазме. От- |
|
ложение керозина происходит в ре- |
|
тикулоэндотелии печени, селезёнки, |
|
костном мозге, лимфоузлах, лёгких. |
|
Чаще болеют женщины. |
Рис. 3.16. Посттравматическая киста селезён- |
Существуют три формы болезни |
ки (макропрепарат удалённой селезёнки) |
Гоше: |
Приведено по: http://www.surgerycom.net/ |
острый раннедетский висцераль |
Galery/Photo.htm |
ный тип(у детей в возрасте до 1 года), |
|
который проявляется быстро прогрес- |
сирующим поражением головного мозга, судорогами, кровоизлияниями, анемией, заканчивается смертью в возрасте около 2 лет;
хронический висцеральнокостный тип у взрослых (с началом заболевания в детстве), который характеризуется длительным, но прогрессирующим течением, ремиссиями (при этом различают два типа – висцеральный и костный;
Рис. 3.17. Внешний вид пациентки, страдающей болезнью Гоше, до (а) и после (б) комбинированного лечения (спленэктомии и применения ферментозаместительной терапии с использо - ванием глюкоцереброзидазы)
Приведено по: http://www.health-ua.com/articles/150.html
120