

Ч 2
.pdf
Синдром (болезнь) Виллебранда (синонимы – синдром Виллебранда– Юргенса, органический тромбопенический синдром, наследственная псевдогемофилия и геморрагический тромбастенический синдром). Забо-
левание, передающееся по наследству. Болеют чаще женщины. В раннем детстве периодически возникают кровотечения из слизистых оболочек, иногда – внутрисуставные кровоизлияния, упорные посттравматические, желудочнокишечные кровотечения, гематурия, кровоизлияния в сетчатую оболочку глаз и кожу. Количество тромбоцитов в норме, но в них имеются небольшие морфологические изменения. Время кровотечения удлинено, свёртываемость и ретракция сгустка в норме.
Описан финским врачом E. A. Willebrand (1870–1949).
Синдром (болезнь) Гланцманна (синонимы – геморрагическая тромбоцитоастения Гланцманна, болезнь Гланцманна–Негеля). Наследственное заболевание, проявляющееся кровотечениями из носа, дёсен, слизистых оболочек. Часты кровоизлияния в кожу (рис. 4.10). Кровотечения сопровождаются анемией. Количество тромбоцитов в норме, но они изменены морфологически (мега- и микротромбоциты). Ретракция сгустка замедлена, увеличена длительность кровотечения, свёртываемость крови в норме.
Описан шведским гематологом E. Glanzmann (1887–1959).
Синдром (болезнь) Де Ври. Врождённое заболевание, носящее семейный характер. Ведущим в клинической картине является геморрагический синдром, сочетающийся с аномалиями развития ладони и синдактилией (между II и III пальцами). При исследовании коагулограммы определяется отсутствие или недостаточная активность фактора V. Течение заболевания благоприятное.
Описан израильским педиатром А. De Vries.
Синдром Казабаха–Меррита. Гигантская гемангиома в сочетании с тромбоцитопенией и reморрагическими кризами. Опухоль багрово-красного цвета, малоболезненна. Периодически возникают геморрагические кризы с кровотечениями в гемангиому, вызывающие её увеличение и напряжение. Развиваются анемия, тромбоцитопения, иногда удлиняется время кровотечения, замедляется время свёртывания крови. Патогенез не изучен.
Рис. 4.10. Длительные, плохо останавливающиеся кровотечения из мелких ранок и царапин (основной клинический признак синдрома Гланцманна)
132
Синдром (болезнь) Коровникова. Сегментарная форма портальной гипертензии, развивающаяся после тромбоза селезёночной вены. Проявляется спленопатией с субтромбоцитозом и гастроэнтероррагиями. Характерны острое начало, обильная кровавая рвота, дёгтеобразный стул. Спленомегалия не выражена. Между кровотечениями отмечают периоды видимого благополучия (иногда несколько лет). При повторных рентгенологических исследованиях пищевого канала патологии не выявляют. Свёртываемость и продолжительность кровотечения близки к норме. Эндотелиальный симптом не постоянен. Отмечаются лейкопения, тромбоцитопения, мегакариоцитоз, субтромбоцитоз. Диагноз уточняют с помощью спленопортографии.
Описан врачом А. Ф. Коровниковым.
Синдром Кошуа–Эппингера–Фругони (синонимы – тромбофлебитическая спленомегалия, синдром Опица). Хронический рецидивирующий тром-
бофлебит воротной вены, проявляющийся периодической лихорадкой, асцитом, гастроинтестинальными кровотечениями, кровоизлияниями в кожу и слизистые оболочки, гепатомегалией и спленомегалией.
Впервые описан австрийским врачом H. Eppinger (1879–1946).
Синдром (болезнь) Крювелье–Баумгартена I. Врождённая гипоплазия воротной вены с атрофией печени без признаков цирроза, явлениями портальной гипертензии и незаращением пупочной вены. При этом постнатально сохраняется эмбриональный вариант воротно-пупочного кровообращения.
Клинически проявляется спленомегалией с явлениями гиперспленизма, развитием коллатерального кровообращения и варикозом вен в трёх сосудистых зонах (вены пищевода и желудка, геморроидальные и околопупочные вены). На животе формируется «голова медузы». Аускультативно выше пупка выслушивают венозный шум, усиливающийся при поднятии пациентом головы и исчезающий при надавливании ладонью выше пупка. Над зоной максимального выслушивания шума пальпаторно отмечают дрожание тканей. Наряду со спленомегалией и кровотечениями отмечаются анемия, лейко- и тромбоцитопения. Рентгенологически и эндоскопически выявляются расширенные вены в нижней трети пищевода. Спленопортография свидетельствует о расширении селезёночной и воротной вен, открытых и резко расширенных пупочной и околопупочной венах. При целиакографии выявляются признаки усиленного артериального притока к селезёнке и желудку.
Описан французским патологоанатомом J. Cruveilhier (1791–1874) и немец-
ким патологоанатомом P. C. Baumgarten (1848–1928).
Синдром (болезнь) Леттерера–Сайве. Нелипоидный ретикулоэндотели-
альный синдром, характеризующийся изменениями в скелете, поражением кожи, гепатоспленомегалией, лимфаденопатией, анемией и склонностью к геморрагиям. Болеют, как правило, дети до 3 лет. Диффузное и прогрессирующее системное поражение проявляется пурпурной, похожей на кровоподтёки, сыпью, гиперплазией дёсен, перемежающейся лихорадкой, прогрессирующей анемией, увеличением печени, селезёнки, лимфатических узлов и прекраще-
133
нием роста. Поражаются кости черепа. При биопсии кожи и лимфоузлов выявляется большое количество ретикулоэндотелиальных клеток, во внутренних органах – скопление гистиоцитов.
Описан в 1952 г. немецким патологоанатомом Е. Letterer и шведским педи-
атром S. A. Siwe.
Синдром Стивена–Джонсона. Эритематозно-экссудативные высыпания на коже, слизистых оболочках пищевого канала и бронхов, приводящие иногда к тяжёлым желудочно-кишечным кровотечениям. Болезнь начинается остро, протекает тяжело. Этиология не известна. Имеются указания на связь с инфекцией, приёмом различных лекарств (барбитуратов, сульфаниламидных препаратов, пенициллинов), гипо- и авитаминозом, коллагенозами. Является разновидностью многоформной экссудативной эритемы.
Описан американскими педиатрами A. N. Stevens (1884–1945) и F. Ch. Johnson (1894–1934).
Синдром (болезнь) Филатова (синонимы – агранулоцитоз, инфекцион-
ный мононуклеоз). Заболевание проявляется некротической ангиной или некрозами дёсен, тонкой или толстой кишки, половых органов, лёгких. Соответственно, выделены три клинические формы – ангинозная, кишечная и лёгочная. Отмечаются общая слабость, высокая температура, дисфагия. Живот мягкий, болезненный, имеется положительный симптом Щёткина–Блюмбер- га, могут быть инфильтраты. В крови лейкопения, нейтропения, лимфоцитоз. Иногда возникает флегмона слепой, тонкой кишок или язвенный колит. Язвы тонкой или толстой кишки могут осложняться кровотечением, перфорацией, перитонитом. В основе заболевания – аллергические реакции. Сенсибилизация может наступить в результате перенесённых инфекционных заболеваний, травм, приёма медикаментов.
Описан педиатром Н. Ф. Филатовым (1847–1902).
Синдром Фишера–Эванса (синоним – синдром Эванса). Выраженная ау-
тоиммунная тромбоцитопения с резкими проявлениями геморрагического синдрома и гемолитической анемии.
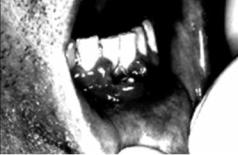
Рис. 4.11. Некротический стоматит при синдроме Франка
Приведено по: http://www.edentworld.ru/cgibin/info/lib.pl?cid=000/&DocID=1410
Описан в 1961 г. американским учёным Е. Н. Fischer и американским гематологом R. S. Evans.
Синдром Франка(синоним – алейкия геморрагическая). Остро проте-
кающеезаболевание,характеризующееся лейкопенией и тромбоцитопенией с последующим развитием анемии. Проявляется геморрагическим диатезом, некротическим стоматитом (рис. 4.11), ангиной, тяжёлым энтероколитом с глубоким поражением стенки кишки.
134

Заболевание может развиться после употребления перезимовавших в поле злаков, после приёма некоторых медикаментов, а также при лучевых поражениях и др.
Описан немецким учёным О. Frank Otto (1865–1944).
Синдром Хегглина (синоним – аномалия Хегглина). Наследственная аномалия состава крови, характеризующаяся тромбоцитопенией, наличием гигантских тромбоцитов, а также нейтрофилов, в которых при окраске азурэозином выявляются включения в виде голубоватых комочков. Иногда сопровождается геморрагическим синдромом. Наследуется по аутосомно-доми- нантному типу.
Описан швейцарским терапевтом R. М. Hegglin (1907–1969).
Синдром (болезнь) Хержманского–Пудлака. Наследственное заболевание человека (тип наследования – аутосомно-рецессивный). Форма тромбоцитопатии, характеризующаяся нарушением структуры и функций тромбоцитов, а также альбинизмом. Имеет место нарушение биогенеза и/или функций субклеточных органелл – меланосом, лизосом и плотных гранул тромбоцитов.
Синдром (болезнь) Шенлейн–Геноха (синонимы – аллергическая пурпура, геморрагический васкулит, геморрагический капилляротоксикоз,
анафилактическая пурпура). Проявляется кожной сыпью, желудочно-ки- шечными симптомами (рис. 4.12) и болезненной припухлостью суставов. Один из признаков может превалировать. Болеют чаще мальчики в возрасте 3–15 лет. На коже нижней части спины, ягодиц, разгибательных поверхностей рук и нижних конечностей в течение 1–2 недель появляется петехиально-па- пулёзная сыпь, вплоть до некроза кожи. Поражение кожи нередко сопровождается кровоизлияниями в пищевой канал (чаще поражается терминальный отдел подвздошной кишки), что приводит к развитию так называемой абдоминальной пурпуры – внезапно появляется схваткообразная боль в области пупка и правой боковой области, реже в надчревной. Одновременно возникают рвота и понос (иногда
спримесью крови). У некоторых пациентов может иметь место запор, возможно развитие гломерулонефрита
сгематурией и альбуминурией. При молниеносной пурпуре кожные про-
явления сочетаются с резко выражен- |
Рис. 4.12. Петехии, эрозии, яркая сосудистая |
ными признаками поражения почек |
|
и пищевого канала. Заболевание мо- |
сетка на слизистой толстой кишки при болез- |
ни Шенлейн–Геноха (данные колоноскопии) |
|
жет осложняться миокардиальным не- |
Приведено по: http://www.colonoscopy.ru/pro- |
крозом. В крови – эозинофилия, свёр- |
jects/marzatka/flat/8.htm |
135

тываемость и длительность кровотечения в норме, тромбоциты без изменений. Природа заболевания аллергическая.
Описан немецкими врачами J. L. Schonlein (1793–1864) и Е. Н. Henoch (1820–1910).
Фактор Стюарта (синонимы – фактор X, Прауэр-фактор, фактор Стю-
арта–Прауэра). Образующийся в печени белок плазмы крови участвует в образовании фактора III свёртывания крови и фактора 3 тромбоцитов. Недостаточность фактора X обусловливает возникновение болезни Стюарта–Прауэра, проявляющейся выраженным геморрагическим синдромом.
Назван по фамилиям пациентов, у которых впервые установлена недостаточность данного фактора.
Фактор Хагемана. Недостаточность фактора XII свёртывания крови, проявляющаяся дефицитом Хагемана и геморрагическим синдромом.
Назван именем пациента, у которого впервые в 1963 г. идентифицирована недостаточность фактора XII свёртывания крови.
4.2. Патология системы гемостаза, сопровождающаяся гиперкоагуляционным синдромом, и её проявления
Симптом Купермана (синоним – контрастное нёбо). Чёткая цветовая граница в месте перехода твёрдого нёба в мягкое. Выявляют при полицитемии.
Синдром (болезнь) Вакеза (синонимы – эритремия, истинная полици-
темия). Болеют в основном лица старше 45–50 лет (чаще мужчины). Клинические признаки: головная боль, головокружение, шум в голове, общая слабость, одышка при движениях, красно-синюшная окраска кожи лица (особенно губ, кончика носа), эритромелалгия, инъецирование сосудов склеры, часто – повышение артериального давления. Возможно желудочно-кишечное кровотече-
ние, обусловленное тромбозом мезентериальных сосудов. Иногда развивается истинный цирроз печени. Могут иметь место тромбозы сосудов печени и селезёнки, тромбоз магистральных сосудов с гангреной конечностей и сосудов мозга. Отмечается полнокровие внутренних органов. Жировой костный мозг вытесняется красным, имеющим полиморфный состав. Ретикулярные волокна разрастаются до степени диффузного фиброза. В селезёнке и печени – очаги миелоидной метаплазии, в крови – значительное увеличение эритро-
Рис. 4.13. Мазок крови при болезни Вакеза |
цитов (до (8–10)1012/л) и |
повышение |
Приведено по: http://www.medslova.ru/index. |
содержания гемоглобина |
(рис. 4.13). |
php?dr=ts/ee&id=eeritremiy.html |
Гематокрит повышается до 60–70 %, |
|
136
выявляются лейкоцитоз и гипертромбоцитоз, гипербилирубинемия.
Заболевание протекает хронически в течение 8–10 лет. В диагностике большую роль играет трепанобиопсия костного мозга.
Описан французским врачом
L. H. Vaquez (1860–1936).
Синдром Моссе. Красная истин-
ная полицитемия, сочетающаяся с циррозом печени. Вначале развивается полицитемия, которая затем осложняется развитием цирроза печени. Увеличение печени умеренное. Заболевание нередко сопровождается тромбозом воротной вены.
Описан в 1907 г. немецким врачом M. Mosse (1873–1936).
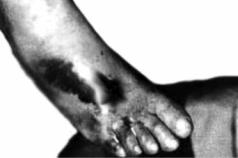
Синдром (болезнь) Нигаарда–Брауна (синоним – эссенциальная тром-
бофилия). Перемежающаяся хромота, тромбозы артерий нижних конечностей с развитием гангрены (рис. 4.14), повышенное содержание фибриногена и глобулинов в сыворотке крови, укороченное время свёртывания крови. В основе заболевания – артериальные тромбозы неясной этиологии.
Описан в 1931 г. американскими врачами К. К. Nygaard и G. E. Brown (1885–1935).
Синдром (болезнь) Пелицеуса–Мерцбахера (синоним – лейкодистро-
фия суданофильная). Врождённое заболевание (форма лейкодистрофии), передающееся по рецессивному (сцепленному с полом) типу. Обусловлено дефектом протеолипидного белка, ген которого (PLP) локализован на участке q21.33–q22 Х-хромосомы. Клинически сопровождается выраженной иммуносупрессией с развитием множественных инфекционных осложнений.
Описан немецкими врачами F. Pelizaeus (1850–1917) и L. Merzbacher (1875– 1942).
Синдром (болезнь) Револя (синонимы – синдром Мортенсена, гипертромбоцитарный миелоз, геморрагическая тромбоцитемия). Сочетание эс-
сенциальной тромбоцитемии и геморрагического диатеза. Заболевание проявляется кровотечениями из носа, гастроинтестинальными кровотечениями, спленомегалией, тромбозами и тромбофлебитами. Отмечают гипопротеинемию, лейкоцитоз, тромбоцитоз (часто значительный) и мегакариоцитоз. Болеют чаще лица пожилого возраста.
Описан французским врачом J. Revole.
137
4.3. Заболевания, сопровождающиеся снижением иммунорезистентности (иммунопатии), и их проявления
В настоящее время под иммунитетом подразумевают защиту организма от всего генетически чужеродного – микробов, вирусов, чужеродных клеток, тканей, хирургически пересаженных органов или генетически изменившихся собственных клеток, включая раковые (М. Г. Сачек с соавт., 1994).
Наука об иммунитете (иммунология) изучает генетические, молекулярные
иклеточные механизмы реагирования организма на чужеродные субстанции, именуемые антигенами, в целях поддержания постоянства его внутренней среды (Р. В. Петров, 1987). Иммунная система в определённых условиях может вызывать патологические процессы по типу аутоиммунных и аллергических реакций. Она не только выполняет защитную функцию, но и оказывает регулирующее действие на другие системы организма. Так, растворимые продукты иммунной системы (медиаторы) являются мощными регуляторами, оказывающими влияние на функцию органов кроветворения, нервную, эндокринную и другие системы. В свою очередь сама иммунная система находится «под контролем» нервной, эндокринной и кроветворной систем.
Действие иммунной системы, заключающееся в распознавании и удалении (элиминации) из организма генетически чужеродного материала, представляет собой сложный многоэтапный процесс – иммунный ответ, или иммунную ре акцию. Иммунный ответ осуществляется специализированными клеточными образованиями – лимфоцитами, многочисленными добавочными клетками
игуморальными факторами иммунитета. Все эти элементы иммунной системы (иммунокомпетентные органы) локализованы преимущественно в крови, лимфе и лимфоидных органах и тканях (костном мозге, вилочковой железе, лимфатических узлах, селезёнке, пейеровых бляшках и лимфоидных фолликулах кишечника, лёгких, коже) и, несмотря на анатомическую разобщённость, связаны между собой в единую систему посредством лимфо- и кровотока.
На генетически чужеродные организму агенты иммунная система реагирует двумя путями – клеточным иммунитетом и гуморальным иммунитетом.
Гуморальный иммунитет – это синтез и секреция В-лимфоцитами и их потомками, а также плазматическими клетками специфических гликопротеидов – антител (относящихся к одному из пяти классов – IgG, IgM, IgA, IgE, IgD), которые циркулируют в кровеносном русле и специфически действуют против генетически чужеродного материала. Такая реакция развивается преимущественно на микроорганизмы, чужеродные эритроциты, белки и подобные им молекулы.
Клеточный иммунитет реализуется путём прямого контакта Т-лим фоцитов с антигенными структурами. Противоопухолевая, противовирусная защита, реакция отторжения трансплантата и ряд других защитных реакций реализуются непосредственно лимфоцитами, называемыми цитотоксическими (т. е. способными разрушать клетки).
138

В красном костном мозге находятся предшественники всех клеток крови и лимфы – полипотентные гемопоэтические стволовые клетки (внешне похожие на лимфоциты клетки диаметром 8 мкм). Они способны образовывать колонии кроветворных и лимфоцитообразующих элементов, каждый из которых является клоном, возникающим из одной клетки (рис. 4.15). Свободно мигри - рующие стволовые клетки костного мозга всегда обнаруживаются в периферической крови. В красном костном мозге (в его миелоидной ткани) из этих клеток формируются клетки-предшественники, из которых путём деления и дифференцировки по трём направлениям образуются форменные элементы крови – эритроциты, лейкоциты и тромбоциты. Из стволовых клеток в красном костном мозге формируются клетки, относящиеся к макрофагальной системе, – моноциты и клетки иммунной системы (В-лимфоциты). Стволовые клеткипереносятсятакжевтимус,гдеонитрансформируютсявТ-лимфоциты.
Все основные иммунологические процессы (в том числе реакции клеточного и гуморального иммунитета) контролируются генетическими факторами. Структурами первичного распознавания антигенов выступают продукты генов, локализованных у человека в коротком плече хромосомы 6 (области главного комплекса гистосовместимости). К таким продуктам относятся антигены гистосовместимости (обозначены у человека как HLA – human leucocytic antigen), располагающиеся на цитоплазматической мембране иммунокомпетентных клеток. В систему HLA входят 5 локусов – А, В, С, D и DR. Антигены локусов А, В и С относят к HLA-антигенам I класса, а антигены
Рис. 4.15. Пути дифференциации клеток иммунной системы
139

локусов D и DR – к HLA-антигенам II класса. HLA-антигены I класса участвуют преимущественно в реакциях трансплантационного иммунитета, HLAантигены II класса регулируют иммунный ответ. При этом распознавание «чужого» иммунокомпетентными клетками осуществляется только в том случае, если это «чужое» комплексуется со «своим», а именно с HLA-II- антигенами цитоплазматической мембраны клеток моноцитарно-макрофа- гального ряда и последующего представления такого комплекса Т-лимфо- цитам. С участием HLA-антигенов II класса реализуются процессы взаимодействия между иммунокомпетентными клетками.
В главном комплексе гистосовместимости особую роль играют так называемые гены иммунного ответа (Ir-гены) II класса, которые контролируют силу иммунного ответа в отношении тимус-зависимых антигенов (реализуется через систему Т-клеток и клеточных взаимодействий).
Важнейшим симптомокомплексом, непосредственно влияющим на результаты лечения хирургических пациентов, служит синдром иммунодефици та. Иммунодефицит – это понижение функциональной активности основных компонентов иммунной системы, ведущее к нарушению защиты организма от микробов и проявляющееся в росте инфекционной заболеваемости. Иммунодефициты делят на первичные (наследственные), обусловленные недоразвитием центральных и периферических органов иммуногенеза, и вторичные (приобрётенные), возникающие в связи с болезнью или проводимым лечением.
Первичный, или наследственный, иммунодефицит (ПИД) – это врож -
дённое нарушение иммунной системы, связанное с генетическими дефектами одного или нескольких её компонентов, а именно: комплемента, фагоцитоза, гуморального и клеточного иммунитета. Общей чертой всех видов ПИД является наличие рецидивирующих хронических инфекций, поражающих различные органы и ткани и, как правило, вызываемых оппортунистическими или условно-патогенными микроорганизмами, т. е. низковирулентной флорой.
В настоящее время идентифицировано более 70 врождённых дефектов иммунной системы, и, вероятно, их число по мере совершенствования методов молекулярной иммунодиагностики будет расти. ПИД – относительно редкие заболевания. Их частота составляет в среднем 1/25 000–1/100 000. Исключением является селективный IgA-дефицит, встречающийся с частотой 1/500– 1/700. Если ориентироваться на данные Европейского регистра первичных иммунодефицитов (отчет ESID за июль 1997*), то отношение числа зарегистрированных больных к населению в среднем по Европе составляет 1:96000, тогда как в отдельных западноевропейских странах – 1:38 000 (Великобритания), 1:25 000 (Нидерланды), 1:12 500 (Швейцария) и даже 1:10 000 (Швеция). Нет оснований считать этнические различия в Европе фактором, существенно влияющим на реальную частоту заболеваний. Различия на порядок можно связать с качеством выявления и активностью регистрации больных.
* Приведено по: Primary Immunodeficiency Diseases: Report of a WHO Scientific Group / F. S. Rosen [et al.] // Clin. and Exp. Immunol. – 1997. – N 109 (Suppl. 1). – P. 1–28.
140