

Ч 2
.pdfРяд сложных врождённых пороков, развившихся вследствие приёма во время беременности различных лекарственных препаратов, приведен на рис. 7.3.
Интоксикации. Рождённые от матерей, употреблявших во время беременности алкоголь, проявляют признаки нарушения умственного и физиче-
Рис. 7.3. Сложные врождённые пороки, обусловленные приёмом лекарств во время беременности: а, б – сиреномелия («синдром русалки» – сросшиеся в виде плавника ноги) в сочетании с недоразвитием желудочно-кишечного тракта и гениталий, отсутствием почек и мочевыводящих путей, пороком сердца; в – сложный порок с отсутствием ушей, аномалией мозга, пищеварительного тракта, сердца; г – сложный порок с экстракардией (расположением сердца вне
грудной клетки); д – циклопизм (один глаз, одно полушарие мозга, сложный порок сердца)
Приведено по: http:// www. foxyhome. ru/index. php? newsid= 1181
221
|
ского развития, которые известны как |
|
|
«алкогольный синдром плода». У жен- |
|
|
щин, курящих во время беременности, |
|
|
возникает повышенный риск выкиды- |
|
|
ша, рождения мертвого ребёнка либо |
|
|
ребёнка с низким весом (последний |
|
|
имеет значительно более высокий шанс |
|
|
стать инвалидом или умереть, чем но- |
|
|
ворождённый с нормальным весом). |
|
|
Недостаточное питание. Само- |
|
Рис. 7.4. Внешний вид новорождённого с врож- |
произвольные аборты, низкий вес при |
|
рождении и другие проблемы связаны |
||
дённым сифилисом (видны инфильтративные |
также с недостаточным питанием ма- |
|
изменения подбородочной области) |
||
тери. |
||
|
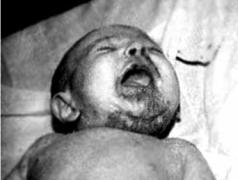
Инфекции. Несмотря на то что плод защищён от многих инфекций, некоторые из них могут приводить к серьёзным дефектам в зависимости от стадии развития плода, во время которой имела место инфекция. Воздействие вируса краснухи на плод вызывает пороки сердца, слепоту, глухоту и другие нарушения. Некоторые инфекции поражают плод до или во время родов, что бывает причиной врождённого заболевания или смерти. Среди них – цитомегаловирусная инфекция и токсоплазмоз (оба часто протекают легко и незаметно для матери), а также болезни, передаваемые половым путём, в частности гонорея, хламидиоз, генитальный герпес и сифилис (рис. 7.4).
Физические факторы внешней среды. Эмбрион или плод может постра-
дать от повышенного уровня ионизирующего излучения (прежде всего это естественный радиоактивный фон и рентгенодиагностика). Хотя считается, что современные методы диагностики не опасны для эмбриона и плода, необходимо закрывать свинцовым фартуком тазовую область у женщин репродуктивного возраста при рентгеноскопии и, если нет чрезвычайных показаний, назначать рентгенологическое исследование через неделю или 10 дней после менструации, так как в этот период беременность маловероятна. Поражающее действие на плод излучения от микроволновых печей, дисплея компьютеров и излучения при проведении диагностического ультразвукового исследования в настоящее время не подтвердилось.
3. Многофакторные причины. Возникновение большинства врождённых пороков нельзя объяснить какой-либо одной генетической причиной или воздействием какого-то одного фактора окружающей среды. Предполагается, что это результат или взаимодействия многих генов (полигенная причинность), или совместного действия генов и факторов окружающей среды (полифакторная причинность).
222
7.1. Пороки развития эндокринной системы, врождённые аномалии обмена веществ
Классификация эпонимических симптомов и синдромов в соответствии с МКБ-10.
Класс IV. Болезни эндокринной системы, расстройства питания и нарушения обмена веществ (E00–E90):
E00 – врождённый синдром йодной недостаточности;
E25 – адреногенитальные расстройства.
Нарушения обмена веществ (E70–E90):
E70 – нарушения обмена ароматических аминокислот;
E71 – нарушения обмена аминокислот с разветвлённой цепью и обмена жирных кислот;
E72 – другие нарушения обмена аминокислот;
E73 – непереносимость лактозы;
E74 – другие нарушения обмена углеводов;
E75 – нарушения обмена сфинголипидов и другие болезни накопления липидов;
E76 – нарушения обмена глюкозаминогликанов;
E77 – нарушения обмена гликопротеинов;
E78 – нарушения обмена липопротеидов и другие липидемии;
E79 – нарушения обмена пуринов и пиримидинов;
E80 – нарушения обмена порфирина и билирубина;
E83 – нарушения минерального обмена;
E84 – кистозный фиброз;
E90 – расстройства питания и нарушения обмена веществ при болезнях, классифицированных в других рубриках.
Отдельные эпонимические симптомы и синдромы.
Анемия Даймонда–Блекфена. Врождённая болезнь неясной этиологии у детей грудного возраста, характеризующаяся недостаточностью эритропоэза и проявляющаяся симптомами анемии.
Описана в 1947 г. американским педиатром L. К. Diamond.
Анемия Фанкони (синоним – синдром Фанкони I). Наследственное заболе-
вание человека с ломкой и рекомбинацией хромосом, которое является одной из форм гипопластической анемии, сочетающейся с тромбоцитопенией и проявляющейся в общем угнетении кроветворных функций костного мозга. Наследуется по аутосомно-рецессивному типу. Нередко отмечаются множественные хромосомные нарушения. Заболевание обусловлено наличием хромосомных аберраций различного типа (как правило, обменов между негомологичными хромосомами), что является следствием нарушения процессов репарации ДНК. Один из вероятных локусов (FA1) сцеплен с длинным плечом хромосомы 20 человека.
В типичных случаях развивается в возрасте 4–6 лет. Довольно часто сочетается с аномалией костной системы (40 %) – недоразвитием лучевой или
223
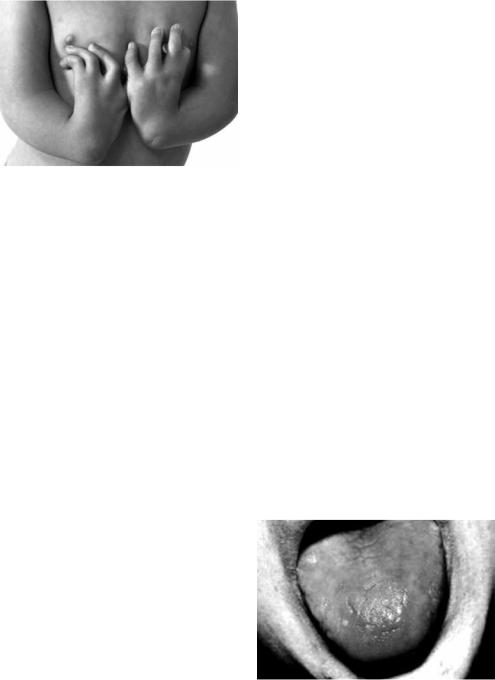
I пястной кости (рис. 7.5), патологической пигментацией кожи (51 %), аномалиями развития внутренних органов (почек, селезёнки). Характерно умственное и половое недоразвитие.
Описана в 1927 г. швейцарским педиатром G. Fanconi.
Анемия Эстрена–Дамешека. На-
следственная гипопластическая анемия у детей с неэффективностью всех трёх ростков кроветворения в костном мозге. Отличается от анемии Фанкони отсутствием других аномалий развития.
Описана в 1951 г. американскими педиатрами S. Estren и W. Dameshek.
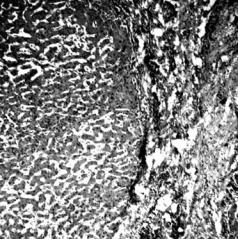
Болезнь Бирмера (синонимы – пернициозная анемия, злокачественная анемия, синдром или анемия Аддисона–Бирмера). Прогрессирующая фор-
ма анемии, возникающая при нарушении усвоения витамина В12. Одной из форм этиологии заболевания является наследственная, связанная с генетически детерминированным недостатком витамин В12-связывающего фактора крови (белка транскобаламина-II: ген TCN2 локализован на длинном плече хромосомы 22). Тип наследования не установлен (рис. 7.6).
Впервые описана немецким врачом A. Biermer (1827–1892).
Болезнь Виллебранда. Наследственное заболевание, обусловленное дефицитом или отсутствием фактора Виллебранда в крови, что приводит к трудно останавливаемым кровотечениям (включая внутренние кровотечения).
Описана финским врачом Е. A. Willebrand (1870–1949).
Болезнь Вильсона (синонимы – Dystrophia hepatocerebralis, болезнь Вестфаля–Вильсона–Коновалова, болезнь Вильсона–Коновалова, гепатолентикулярная дегенерация, лен-
тикулярная прогрессирующая деге-
нерация). Наследственно детерминированная гепатоцеребральная дистрофия, характеризующаяся нарушением экскреции ионов меди с желчью и их включения в церулоплазмин. Сопровождается поражением печени (гепа-
тит с формированием телец Меллори, |
Рис. 7.6. Характерный вид языка при анемии |
жировая дистрофия, цирроз) (рис. 7.7) |
Аддисона–Бирмера |
и дистрофическими процессами в го- |
Приведено по: http://img.tfd.com/mosby/ |
ловном мозге (в чечевидном ядре |
thumbs/500038-fx18.jpg |
224
и бледном шаре, в больших полушариях и мозжечке). Наследуется по ау- тосомно-рецессивному типу (локус WND расположен на участке q14.2– q21 хромосомы 13). Встречается чаще у мужчин в возрасте 10–25 лет.
Ранние признаки болезни – гепато- и спленомегалия, желтуха, анорексия. Среди неврологических проявлений преобладают тремор, повышенный тонус мышц с переходом
вригидность, адинамия (с исходом
вконтрактуры и обездвиженность), гиперкинезы (типа атетоза) и клони-
ко-тонические судороги, изменение речи, снижение памяти и интеллекта (вплоть до деменции). Отмечаются также отложения жёлто-бурого пигмента на роговицах (кольца Кайзе- ра–Флейшнера), застывшая улыбка,
кожный зуд, артропатии, диффузный остеопороз, нарушения кровообращения, ведущие к гипоксии. Выявляются гипопротеинемия, гемолитические кризы, аминоацидурия и гиперкупроурия (до 1000 j в сутки). Иногда отмечаются желтовато-коричневая пигментация кожи лица и туловища, геморрагический диатез, желтуха, кожный зуд. Портальная гипертензия с желудочным кровотечением развивается в поздние периоды болезни. Нередки случаи смерти от печёночной комы. Прогноз в большинстве случаев неблагоприятный.
Описана в 1912 г. английским невропатологом А. К. Wilson (1878–1937) и невропатологом Н. В. Коноваловым (1900–1966).
Болезнь Вольмана. Наследственная болезнь, обусловленная недостатком выработки лизосомальной кислой эстеразы и накоплением в клетках организма триглицеридов и эфиров холестерина. Наследуется по аутосомно-рецес- сивному типу. Проявляется рвотой сразу после рождения ребёнка, частым жидким стулом, отставанием в развитии (в первую очередь дефицитом массы тела). Со стороны пищеварительного тракта отмечаются гепато- и спленомегалия, значительное увеличение размеров живота, множественный ксантоматоз кожи, увеличение надпочечников с их кальцификацией (последние становятся видны при рентгенографии). При гистологическом исследовании в печени, селезёнке, надпочечниках и костном мозге находят пенистые клетки со значительным содержанием триглицеридов и эфиров холестерина.
Описана в 1961 г. американским педиатром М. Wolman.
Болезнь Галлервордена–Шпатца (синоним – прогрессирующая ригид-
ность). Наследственная болезнь, связанная с нарушением обмена железа и ли-
225
пидов. Наследуется по аутосомно-рецессивному типу. Проявляется в возрасте 7–9 лет повышением мышечного тонуса, гиперкинезами, прогрессирующей деменцией, снижением зрения и пигментным ретинитом.
Впервые описана немецким невропатологом J. Hallervorden (1882–1965).
Болезнь (синдром) Герса (синонимы – гликогеноз VI типа , гепатофосфорилазная недостаточность, синдром Херса). Гликогеноз, обусловленный недостаточностью гликоген-фосфорилазы печени. Наследуется по аутосомнорецессивному типу (ген PYGL локализован на участке q11.2-q24.3 хромосомы 14). Характеризуется отставанием в росте, выраженной гепатомегалией с гиперхолестеринемией, умеренной гипогликемией, липемией, гипогликемией, лактатным ацидозом. Прогноз может быть благоприятным (при компенсации нарушений гликогена глюконеогенезом).
Описана в 1958 г. бельгийским биохимиком H. G. Hers.
Болезнь Гланцманна–Негели (синонимы – тромбацитастения Гланцманна, геморрагическая тромбастения). Наследственная болезнь из группы геморрагических диатезов, характеризующаяся недостаточностью ряда ферментов в тромбоцитах, вторичным нарушением ретракции кровяного сгустка, а зачастую удлинённым временем кровотечения при нормальном или слегка пониженном количестве тромбоцитов, наличием гигантских тромбоцитов. Выделяют формы с аутосомно-рецессивным и аутосомно-доминантным типом наследования.
Описана швейцарскими гематологами Е. Glanzmann (1887–1959) и О. Naegeli (1871–1938).
Болезнь Гринфилда (синонимы – метахроматическая лейкодистрофия, лейкодистрофия Гринфилда–Шольца, поздняя инфантильная лейкоди-
строфия). Является наследственным заболеванием человека, в основе которого – врождённый дефицит активности лизосомной арилсульфатазы А. Наследуется по аутосомно-рецессивному типу (ген ARSB локализован на участке q13.31–qter хромосомы 22). Выделяют три формы, различающиеся по времени проявления, что зависит от уровня остаточной активности мутантного фермента. После развития заболевания, как правило, быстро наступает летальный исход из-за необратимой атрофии головного мозга. Болезнь проявляется в раннем детском возрасте гиперкинезами, параличами, задержкой психического развития, повышенным содержанием сульфатидов в цереброспинальной жидкости, крови и моче.
Описана английским невропатологом J. G. Greenfield (1884–1958).
Болезнь Гюнтера (синонимы – эритропоэтическая уропорфирия, lroporphyria erythropoetica, врождённая порфирия). Наследственная порфирия,
сопровождающаяся врождённой спленомегалией и гепатомегалией (рис. 7.8). Клинически проявляется до 5-летнего возраста – моча становится ярко-розо- вого или красного цвета, развивается гиперчувствительность к свету (на открытых частях тела возникают везикулы или буллы, часто изъязвляющиеся и заживающие через рубцевание с формированием участков гиперпигмента-
226
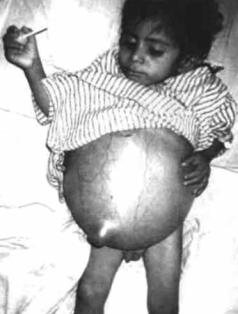
ции или депигментации). Важный клинический признак – спленомегалия с гиперспленизмом, проявлением которого является рецидивирующий гемолиз, сочетающийся с повышением уробилина в кале. Зубы часто окрашены в жёлто-коричневый или фиолетовый цвет. В моче и крови определяется повышенная концентрация уропорфирина (моча приобретает красный цвет).
Описана немецким терапевтом Н. Gunther (1884–1956).
Болезнь Зейтельбергера. Наслед- |
|
|
ственное заболевание, характеризую- |
|
|
щееся нарушением обмена веществ |
|
|
с прогрессирующей липоидной деге- |
|
|
нерацией в центральной нервной си- |
|
|
стеме, что приводит к амавротической |
Рис. 7.8. Внешний вид ребёнка, страдающего |
|
идиотии и другим последствиям. На- |
||
следуется по аутосомно-рецессивному |
болезнью Гюнтера |
|
Приведено по: http://www.uranmunition.net/ |
||
типу (обусловлено дефектом фермен- |
||
images1/morbus_guenther.jpg |
та-N-ацетилгалактозаминидазы, ген
NAGA локализован на участке q13–qter хромосомы 22). Описана немецким учёным Н. Seidlberger.
Болезнь (синдром) Кори (синонимы – гликогеноз III типа , лимитдек-
стриноз, болезнь Форбса). Гликогеноз, обусловленный врождённым полным или частичным отсутствием активности амино-1,6-глюкозидазы и/или глико- ген-ветвящего фермента в мышцах и печени. Характеризуется гепатомегалией, мышечной гипотонией, гипертрофией отдельных мышечных групп, нарушениями сердечной проводимости и кровообращения.
Описана американским биохимиком G. Th. Cori (1896–1957) и американ-
ским хирургом W. S. Forbes (1831–1905).
Болезнь (синдром) Костманна (синоним – агранулоцитоз). Наследствен-
ное заболевание человека, характеризующееся аномалиями крови (эозинофилией и др.) и костного мозга. Наблюдается у грудных детей при неблагоприятном прогнозе. Наследуется по аутосомно-рецессивному типу. Агранулоцитоз обусловлен дефицитом фактора дифференциации нейтрофилов (локус NDF расположен на участке p21.3 хромосомы 6).
Описана в 1961 г. швейцарским педиатром R. Kostmann.
Болезнь Краббе–Бенеке (синонимы – глобоидно-клеточная лейкодистрофия, диффузный инфантильный склероз Краббе). Наследственная бо-
лезнь, обусловленная нарушением синтеза цереброзидов и миелинизацией
227
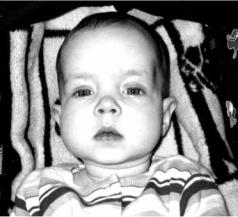
нервных волокон. Проявляется в детском возрасте судорогами, спастическими параличами, деменцией, слепотой и тугоухостью (рис. 7.9).
Впервые описана датским невро-
патологом К. Н. Krabbe (1885–1961).
Болезнь (синдром) ЛеттерераСиве (синонимы – болезнь Абта–Лет- терера–Сиве, нелипоидный ретику-
логистиоцитоз). Острый системный прогрессирующий наследственный гистиоцитоз у детей 1–2 лет, проявляющийся лихорадкой, гепатоспленомегалией, генерализованным увеличением лимфатических узлов, папулёзносквамозной сыпью, геморрагической пурпурой, поражением слизистых оболочек, плоских костей и лёгких.
Описана в 1952 г. немецким патологоанатомом Е. Letterer и шведским пе-
диатром S. A. Siwe.
Болезнь Ли I. Наследственное заболевание человека (наследственная энцефаломиопатия в детском возрасте), характеризующееся повышенной восприимчивостью к инфекциям, мышечной слабостью, гипотонией и другими симптомами. Обусловлена мутацией в митохондриальном геноме (вероятно, заменой тимина на гуанин в положении 8993, что ведет к замене аргинина на лейцин в составе 6-й субъединицы фермента АТФазы).
Описана в 1951 г. английским невропатологом D. Leigh.
Болезнь Мак-Ардла (синонимы – гликогеноз V типа , болезнь Мак- Ардла–Шмида–Пирсона, миофосфорилазная недостаточность, болезнь В. MacArdle). Гликогенез, обусловленный недостаточностью мышечной гли- коген-фосфорилазы. Наследуется по аутосомно-рецессивному типу. Проявляется слабостью и тугоподвижностью мышц, мышечными спазмами, тахикардией. В венозной крови понижено содержание лактата и пирувата, может наблюдаться гемоглобинурия.
Описана английским педиатром В. MacArdle, а также американскими вра-
чами R. Schmid и С. М. Pearson.
Болезнь Оврена (синонимы – гипоакцелеринемия, парагемофилия).
Наследственная болезнь из группы геморрагических диатезов, обусловленная недостаточностью фактора V свёртывающей системы крови. По клиническим проявлениям аналогична гемофилии (основные клинические проявления связаны с выраженным гипокоагуляционным синдромом).
Описана в 1947 г. норвежским гематологом P. A. Owren.
228
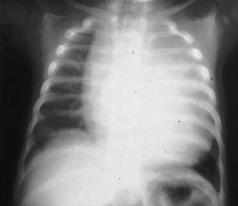
Болезнь Олперса. Наследственная болезнь, обусловленная недостаточностью комплекса I дыхательной цепи митохондрий. Для неё характерны рано появляющиеся эпилептические припадки, деменция, спастичность, слепота и нарушение функции печени. Прогноз в большинстве случаев неблагоприятный.
Болезнь (синдром) Помпе (синонимы – гликогеноз VI типа, генерализо-
ванный гликогенез). Врождённое заболевание, возникающее в связи с дефицитом фермента кислой экзо-1,4-альфа-глюкозидазы, участвующей в метаболизме гликогена. Наследуется по аутосомно-рецессивному типу (ген GAA локализован на участке q23 хромосомы 17).
Болезнь проявляется через несколько дней (недель) после рождения. Отмечаются расстройства дыхания, цианоз, беспокойство, апатия, адинамия, мышечная слабость и микроглоссия. Развиваются гепатомегалия, гипертрофия языка, пилороспазм, отёки, кардиомегалия, бронхиты и гипостатическая пневмония. Имеет место явная задержка роста. В органах и тканях наблюдается избыточное отложение гликогена. В сыворотке крови повышено содержание мочевой кислоты, глутамин-щавелево-уксусной трансаминазы и альдолазы. Сопровождается выраженной мышечной слабостью и отсутствием прибавки в весе (уже в первые недели жизни, а при некоторых формах заболеваниях и позже, масса тела ребёнка перестаёт увеличиваться). Возможны также замедление умственного развития и повторяющиеся респираторные инфекции. Гликоген накапливается в сердце (рис. 7.10), мышцах, центральной нервной системе, печени, почках и надпочечниках. Прогноз неблагоприятный – заболевание заканчивается смертью на первом году жизни от сердечной недостаточности и инфекции (юношеская и взрослая формы заболевания отмечаются крайне редко).
Описана в 1932 г. голландским врачом J. С. Pompe.
Болезнь (синдром) Сандгоффа
(синоним – GM2-gangliosidosis II). На-
следственное заболевание, характеризующееся резкими неврологическими отклонениями, обусловленными мутацией в составе гена-субъединицы гексозаминидазы А и В, которая приводит к аномальному отложению гиглиозида Gm-2 (преимущественно в нервной системе и внутренних органах). Типичным признаком болезни является отставание в психомоторном развитии в первые 6 мес. жизни. На глазном дне определяется симптом «вишнёвой косточки». Развивается ге-
патоспленомегалия, а при прогрессировании заболевания отмечаются миоклонические судороги и спастический тетрапарез. Смерть наступает в 2–3 года. Наследуется по аутосомно-рецессивному типу (ген HEXB локализован на участке q13 хромосомы 5).
Описана в 1968 г. врачом К. Sandhoff.
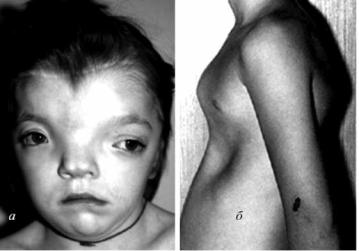
Болезнь (синдром) Слая (синоним – мукополисахаридоз VII типа). На-
следственное заболевание человека с аутосомно-рецессивным типом наследования. Обусловлено дефицитом бета-D-глюкуронидазы (в первую очередь в лейкоцитах и кожных фибробластах), проявляется аномальным накоплением хондроитинсульфатов в соединительной ткани (ген GUSB локализован на участке q22 хромосомы 7).
Характеризуется черепно-лицевыми аномалиями (грубые черты лица с гипертелоризмом, запавший нос, вывернутые вперёд ноздри), поражениями костей и суставов (низкий рост, килевидная грудная клетка, тораколюмбальный кифоз, косолапость), гепатомегалией, спленомегалией, паховыми и пупочными грыжами, помутнением роговицы, фиброзно-мышечной дисплазией аорты (приводящей к сердечной недостаточности), рецидивирующими лёгочными инфекциями и др. (рис. 7.11). Существует клиническая форма заболевания, при которой отсутствует гепатоспленомегалия – имеется небольшое помутнение роговицы, фиброзно-мышечная дисплазия аорты с инфильтрацией мукополисахаридами, приводящая к аортальной регургитации и сердечной недостаточности. Рентгенологически выявляются клювовидные позвонки, расширенные длинные трубчатые кости, множественный дизостоз.
Описана в 1973 г. врачом W. Sly.
Рис. 7.11. Внешний вид пациента с синдромом Слая: а – изменения со стороны лицевого черепа; б – типичные ортопедические составляющие синдрома (низкий рост, килевидная грудная клетка, тораколюмбальный кифоз)
Приведено по: http://belchenko.ru/img/03/10.jpg, http://www.rusmedserv. com/pedthorsurg/cardio1.jpg
230