

Ч 2
.pdfРис. 7.62. Внешний вид и рентгенограмма башнеобразного черепа при синдроме Терсиля (стрелками обозначен феномен «пальцевых вдавлений»)
Приведено по: http://www.ordodeus.ru/Ordo_Deus12_Bashennyj_cherep. html, http://www. myneuro.ru/24/24_2/
Синдром Фара (синоним – наследственное обызвествление мозговых сосудов). Наследственная болезнь, характеризующаяся обызвествлением сосудов, снабжающих внутреннюю капсулу и базальные ядра головного мозга. Тип наследования не установлен. Проявляется в возрасте 20–30 лет атетозом, тремором, центральными параличами, эпилептиформными припадками и прогрессирующим слабоумием.
Описан немецким патологоанатомом К. Th. Fahr (1877–1945).
Синдром Фуа–Иллемана. Врождённое сочетание аплазии крестцовой и копчиковой костей с дисплазией нижних сегментов спинного мозга. Проявляется нижней периферической параплегией с нарушением функций органов таза.
Описан французскими врачами Ch. Foix (1882–1927) и P. Hillemand (1895– 1949).
Синдром Хермана–Агилар–Закса. Наследственная аномалия (аутосом- но-доминантное наследование) – сочетание тугоухости или полной глухоты вследствие аномалий развития внутреннего уха и сахарного диабета с периферической нейропатией. Проявляется периферической нейропатией, диабетической нефропатией или пиелонефритом и ранним старческим слабоумием. В крови выявляются гипергликопротеидемия, гипермукопротеидемия, в моче – гипераланинурия, гиперлейцинурия и гипервалинурия.
Описан в 1967 г. американскими врачами Ch. Jr. Herrman, M. J. Aguilar и O. W. Sacks.
Синдром (болезнь) Шарко–Мари–Тута (синонимы – амиотрофия Шар- ко–Мари–Тута, наследственная невральная амиотрофия). Наследственно детерминированная патология (форма амиотрофии). Характеризуется поражением периферических нервов дистальных отделов конечностей (рис. 7.63) и соответствующими нарушениями чувствительности и параличами. Насле-
291
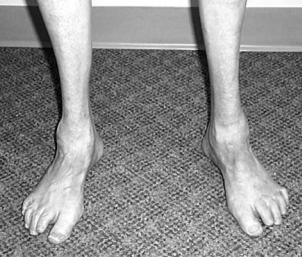
Рис. 7.63. Внешний вид нижних конечностей при синдроме Шарко–Мари–Тута Приведено по: http://www.myfootshop.com/images/medical/ortho/cmt1.jpg
дуется по аутосомно-доминантному (редко – по аутосомно-рецессивному) типу. Известно несколько генетически дифференцированных форм (локус СМТ1A расположен на участке р11.2 хромосомы 17, локус CMT1B – на длинном плече хромосомы 1, СМТХ – на длинном плече Х-хромосомы (q11–q13)).
Описан французскими невропатологами J. М. Charcot (1825–1893), P. Marie (1853–1940) и Н. Н. Tooth (1856–1925).
СиндромШтрюмпелля (синонимы –спастическаяпараплегияШтрюм- пелля, болезнь Эрба–Шарко–Штрюмпелля). Хроническая прогрессирую-
щая наследственно-семейная болезнь нервной системы, характеризующаяся развитием центрального нижнего парапареза при сохранности функций тазовых органов. Наследуется по аутосомно-рецессивному и доминантному типу.
Описан немецким невропатологом A. G. G. Strumpell (1853–1925).
Хорея Гентингтона (синонимы – наследственная хорея, хорея Хантингтона, дегенеративная хорея, хроническая прогрессирующая хорея). На-
следственное заболевание человека с поражением головного мозга. Наследуется по аутосомно-доминантному типу (локус HD расположен на участке p16.3 хромосомы 4). Встречается с частотой 4–560 случаев на 100 000 населения. Генетическое консультирование при диагностике имеет ограниченную ценность. Диагноз ставится на основании ДНК-анализа.
Заболевание протекает с преимущественным поражением мелких клеток полосатого тела и атрофией коры головного мозга, проявляющимися хореическими и атетоидными гиперкинезами в сочетании с нарастающей деменцией. Нередко начинается с изменения личности: человек становится апатичным, начинает пренебрежительно относиться к самому себе, в отдельных случаях развивается психоз. Позже присоединяются неуклюжесть движений, стран-
292
ная суетливость, по лицу проплывают какие-то гримассы (это может быть ошибочно принято за манерность), а вслед за этим проявляются гемихорея и ригидность. Память относительно сохранена. Познавательные способности резко снижены. На ЭЭГ – исчезновение или ослабление a-ритмов.
Описана американским врачом G. S. Huntington (1851–1916).
7.3. Врождённые аномалии (пороки развития) глаза, уха, лица и шеи
Классификация эпонимических симптомов и синдромов в соответствии с МКБ-10:
Q10 – врождённые аномалии (пороки развития) века, слёзного аппарата и глазницы;
Q11 – анофтальм, микрофтальм и макрофтальм;
Q12 – врождённые аномалии (пороки развития) хрусталика;
Q13 – врождённые аномалии (пороки развития) переднего сегмента глаза; Q14 – врождённые аномалии (пороки развития) заднего сегмента глаза; Q15 – другие врождённые аномалии (пороки развития) глаза;
Q16 – врождённые аномалии (пороки развития) уха, вызывающие нарушение слуха;
Q17 – другие врождённые аномалии (пороки развития) уха;
Q18 – другие врождённые аномалии (пороки развития) лица и шеи.
Отдельные эпонимические симптомы и синдромы.
Болезнь Диммера (синонимы – болезнь Бибера, дистрофия Гааба , ре-
шётчатая дистрофия роговицы). Семейно-наследственная болезнь, характеризующаяся появлением в строме оптической зоны роговицы нитевидных, линейных, ветвящихся и переплетающихся между собой помутнений серого цвета (рис. 7.64). Начинается в детском и юношеском возрасте.
Впервые описана австрийским офтальмологом F. Dimmer (1855–1926). Болезнь Норри. Наследственное
заболевание человека, характеризующееся ранней дисплазией сетчатки, наличием в области стекловидного тела опухолевидных масс серо-жел- товатого цвета, развитием увеита, катаракты, вторичной глаукомы и атрофии глазных яблок с полной утратой зрения. Часто сочетается с умственной отсталостью, глухотой и др. Наследуется по рецессивному, сцепленному с полом, типу (локус NDP рас-
положен на участке p11.23–p11.3 X-хро- |
Рис. 7.64. Помутнение роговицы, видимое на |
мосомы). |
щелевой лампе, при болезни Диммера |
Описана в 1899 г. датским офталь- |
Приведено по: http://mega.km.ru/ HEALTH/ |
мологом G. Norrie. |
Preview/oft0018p.jpg |
293
Болезнь Огути. Семейно-наследственная болезнь, проявляющаяся гемералопией, нарушением цветового зрения и своеобразным обесцвечиванием глазного дна с временным восстановлением нормальной окраски после пребывания в темноте.
Описана японским офтальмологом C. Oguchi (1875–1945).
Синдром Аксенфельда. Аномалия развития – сочетание колобомы радужки (реже – аниридии), помутнения хрусталика и эктопии зрачка. Наследуется по аутосомно-доминантному типу.
Описан немецким офтальмологом К. Th. Axenfeld (1867–1930).
Синдром Петерса. Наследственная аномалия развития – дефект задней поверхности роговицы с центрально расположенным бельмом, сопровождающийся уплощением передней камеры глаза. Часто сочетается с другими аномалиями развития, например с волчьей пастью, атрезией кишечника. Предположительно наследуется по аутосомно-рецессивному типу.
Впервые описан немецким офтальмологом A. Peters (1862–1938). Синдром Ригера. Комплекс наследственных аномалий у человека (разви-
тие вторичной глаукомы и другие поражения глаз, дисплазия зубной эмали и т. д.). Наследуется по аутосомно-доминантному типу (локус RGS расположен на участке q22.2–q25 хромосомы 4).
Описан в 1932 г. австрийским офтальмологом Н. Rieger.
Синдром Фера (синонимы – дистрофия роговицы Фера, пятнистая дис-
трофия роговицы). Наследственная болезнь глаз, характеризующаяся постепенно прогрессирующим помутнением роговиц с появлением на них сероватых непрозрачных пятен и снижением остроты зрения. Наследуется по ауто- сомно-рецессивному типу.
Описан в 1902 г. немецким офтальмологом О. Fehr.
Синдром Штаргардта (синоним – юношеская дегенерация жёлтого пят-
на Штаргардта). Семейно-наследственная двусторонняя дегенерация сетчатки, развивающаяся в возрасте 8–15 лет и характеризующаяся поражением жёлтого пятна (пятна сетчатки). Проявляется снижением остроты зрения, развитием центральной скотомы. Наследуется по аутосомно-рецессивному типу.
Описан немецким офтальмологом К. В. Stargardt (1875–1927).
Синдром Штиллинга–Тюрка–Дуэйна (синонимы – синдром Дуэйна, синдром ретракции глазного яблока). Врождённый, чаще односторонний,
паралич прямых мышц глазного яблока, проявляющийся резким нарушением его движений и оттягиванием глаза кзади (ретракцией). Часто сочетается с другими аномалиями развития.
Описан немецким офтальмологом J. Stilling (1842–1915), швейцарским офтальмологом S. Turk и американским офтальмологом A. Duane (1858–1926).
Узелковая дистрофия роговицы Греноува. Наследственная болезнь, ха-
рактеризующаяся дистрофией роговицы с развитием мелких серых узелков в различных её слоях. Наследуется по аутосомно-доминантному типу.
Описана немецким офтальмологом A. Grenouw (1862–1945).

Глава 8
ВРОЖДЁННЫЕ АНОМАЛИИ СИСТЕМЫ КРОВООБРАЩЕНИЯ, ОРГАНОВ ДЫХАНИЯ, ЧЕЛЮСТНО-ЛИЦЕВОЙ ОБЛАСТИ И СИСТЕМЫ ПИЩЕВАРЕНИЯ
«Генетический фонд является наиболее ценным благом человечества. Он определяет жизнь нашего потомства и здоровое, гармоничное развитие грядущих поколений. Будучи экспертами, мы утверждаем, что здоровье будущих поколений находится под угрозой возрастающего развития атомной индустрии и роста количества источников излучения: Мы также считаем, что факт появления новых мутаций у людей является роковым для них и для их потомков».
М. Мuller, эксперт ВОЗ, лауреат Нобелевской премии (1956)
8.1. Врождённые аномалии (пороки развития) системы кровообращения
Классификация эпонимических симптомов и синдромов в соответствии с МКБ-10:
Q20 – врождённые аномалии (пороки развития) сердечных камер и соединений;
Q21 – врождённые аномалии (пороки развития) сердечной перегородки; Q22 – врождённые аномалии (пороки развития) лёгочного и трёхстворча-
того клапанов;
Q23 – врождённые аномалии (пороки развития) аортального и митрального клапанов;
Q24 – другие врождённые аномалии (пороки развития) сердца; Q25 – врождённые аномалии (пороки развития) крупных артерий; Q26 – врождённые аномалии (пороки развития) крупных вен;
Q27 – другие врождённые аномалии (пороки развития) системы периферических сосудов;
Q28 – другие врождённые аномалии (пороки развития) системы кровообращения.
Отдельные эпонимические симптомы и синдромы (основные эпонимиче-
ские синдромы и симптомы, касающиеся хирургической патологии сердца, см. в т. 1, главе 11 «Хирургические заболевания сердца»).
Болезнь Ван-Богарта–Диври (синоним – кортикоменингеальный диф-
фузный ангиоматоз). Наследственная болезнь из групппы ангиоматозов, характеризующаяся врождённой пойкилодермией с телеангиэктазиями, неврологическими и психическими нарушениями (судорожными припадками, экс-
295
трапирамидными расстройствами, деменцией). Наследуется по рецессивному, сцепленному с полом, типу.
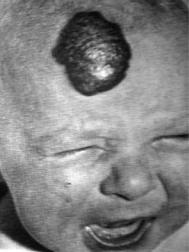
Описана в 1931 г. бельгийскими невропатологами L. Van Bogaert и P. Divry. Синдром Бина. Врождённая ангиопатия с аутосомно-доминантным типом наследования. На коже головы (рис. 8.1), конечностей и тела (иногда на слизистых оболочках пищеварительного тракта) отмечаются синюшные ка-
вернозные гемангиомы, склонные к кровотечению. Описан врачом W. B. Bean.
Синдром Клиппель–Треноне (синоним – варикозный остеогипертро-
фический невус). Врождённое заболевание, характеризующееся гемангиоэктазиями с наличием варикозно расширенных вен нижних конечностей (иногда поражаются и верхние конечности). Причина синдрома – аномалия развития (дисплазия) глубоких вен.
Описан французскими врачами М. Klippel (1858–1942) и P. Trenaunay (1875–1956).
Синдром Ланнелонга–Ашара. Врождённая слоновость. В основе заболевания – врождённое расширение лимфатических сосудов с последующим лимфостазом.
Впервые описана французским врачом Е. Ch. Achard (1860–1944).
Синдром (болезнь) Милроя (синонимы – лимфодерма, врождённая семейная слоновость, болезнь Милроя–Мейжа, синдром Нонне–Милроя– Мейжа). В основе заболевания – врождённая эктазия, гипоплазия или аплазия
лимфатических сосудов (рис. 8.2). Описаны случаи семейного поражения |
||
|
с доминантным типом наследования. Болеют |
|
|
чаще женщины. На начальных стадиях воз- |
|
|
никает отёчность, а затем тотальное заме- |
|
|
щение подкожной основы грубоволокнистой |
|
|
соединительной тканью. Основным проявле- |
|
|
нием болезни является отёк лица, кистей |
|
|
и нижних конечностей (голеней). Отёки обна- |
|
|
руживают при рождении ребёнка или через |
|
|
несколько месяцев. Вначале отёк «мягкий», |
|
|
уменьшается во время сна, однако после на- |
|
|
грузки вновь нарастает. При прогрессирова- |
|
|
ниии процесса «мягкая» (отёчная) стадия пе- |
|
|
реходит в «твёрдую» (склеротическую). При |
|
|
этом ткани становятся плотными, кожа гру- |
|
|
бой, неподвижной, углублений от вдавлений |
|
|
не возникает. Постоянные трофические рас- |
|
Рис. 8.1. Врождённая кавернозная |
стройства – гиперкератоз, трещины, пигмен- |
|
тация, папилломатозные разрастания, иногда |
||
гемангиома в области лба при син- |
||
дроме Бина |
можно наблюдать отёки лёгких, гортани, пи- |
|
Приведено по: Н. Б. Ситковский |
щевого канала с соответствующей клиникой. |
|
с соавт. (1981) |
Гистаминовая проба даёт бурную реакцию. |
|
296
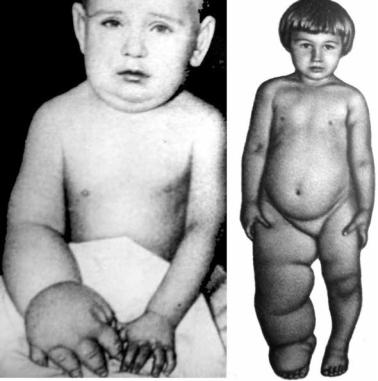
Рис. 8.2. Врождённый лимфостаз руки и ноги (семейная гипоплазия/аплазия лимфатических сосудов) при болезни Милроя
Приведено по: Н. Б. Ситковский с соавт. (1981)
Впервые описан американским врачом W. F. Milroy (1855–1942).
Синдром Паркса–Вебера–Рубашова (синонимы – остеогипертрофический варикозный невус, гипертрофическая гемангиэктазия). Врождённое заболевание, характеризующееся наличием артерио-венозных коммуникаций. Иногда поражаются не только ноги, но и другие органы (печень, почки и т. д.). Характерна триада симптомов – гипертрофия конечности (гигантизм), варикозное расширение вен и гемангиоэктазии, над которыми выслушивается систолический шум. Конечность удлиняется и увеличивается в объёме, кожа уплотняется, развиваются гипертрихоз, гиперкератоз, язвы, тромбофлебиты, могут быть кровотечения. Болеют чаще лица женского пола.
Впервые описан английским врачом F. Parkes Weber (1863–1962). Синдром (болезнь) Пратта. Врождённые мелкие артериовенозные со-
устья нижних конечностей.
Описан в 1949 г. американским хирургом О. Н. Pratt.
Синдром Рандю–Ослера (синонимы – болезнь Ослера–Рандю, болезнь Рандю–Вебера–Ослера, геморрагический ангиоматоз, наследственная геморрагическая ангиома, наследственная геморрагическая телеангиэкта-
297
зия). Множественные наследственные телеангиэктазии, сопровождающиеся повторными кровотечениями. Основной симптом – кровотечение, характер, частота и интенсивность которого зависят от величины, локализации и количества телеангиэктазий. Кровотечения из поражённых опухолью мест возникают спонтанно или после травмы. Наиболее часты носовые кровотечения, но могут быть и внутренние (из пищевого канала, лёгких, почек). Этиология не известна. Гистологически определяются синусообразные расширения сосудов, в стенке которых сохранён один эндотелий. Возникновение телеангиэктазий с последующим кровотечением обусловливает врождённая слабость мезенхимы.
Описан французским врачом Н. J. L. М. Rendu (1844–1902), канадским вра-
чом W. Osler (1849–1919) и английским врачом F. P. Weber (1863–1962).
Синдром Романо–Уорда (синоним – long-QT syndrome). Наследственное заболевание человека, характеризующееся аномалиями функций сердца (асинхронная реполяризация миокарда). На электрокардиограмме отмечается значительное удлинение интервала QT (в основном у детей и молодых людей). Наследуется по аутосомно-доминантному типу (локус LQT расположен на участке p21.3 хромосомы 6).
Впервые описан итальянским врачом С. Romano (1847–1913).
8.2. Врождённые аномалии (пороки развития) органов дыхания
Классификация эпонимических симптомов и синдромов в соответствии с МКБ-10:
Q30 – врождённые аномалии (пороки развития) носа;
Q31 – врождённые аномалии (пороки развития) гортани;
Q32 – врождённые аномалии (пороки развития) трахеи и бронхов;
Q33 – врождённые аномалии (по-
|
роки развития) лёгкого; |
|
Q34 – другие врождённые анома- |
|
лии (пороки развития) органов дыхания. |
|
В клиническом плане наибольшее |
|
распространение получила следую- |
|
щая классификация врождённых ано- |
|
малий дыхательной системы: |
|
1. Аномалии, обусловленные ком- |
|
бинированными нарушениями разви- |
|
тия нескольких структур: агенезия |
|
лёгкого или доли (аплазия лёгкого |
|
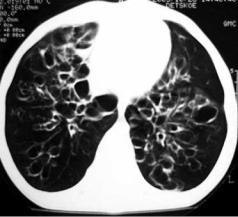
или доли), гипоплазия лёгкого или |
Рис. 8.3. Врождённая буллёзная эмфизема обоих |
доли (простая, кистозная, в том числе |
лёгких (данные компьютерной томографии) |
поликистоз) (рис. 8.3, 8.4), добавоч- |
Приведено по: http://nld.by/200/stat8_1b.jpg |
ные доли лёгкого. |
298
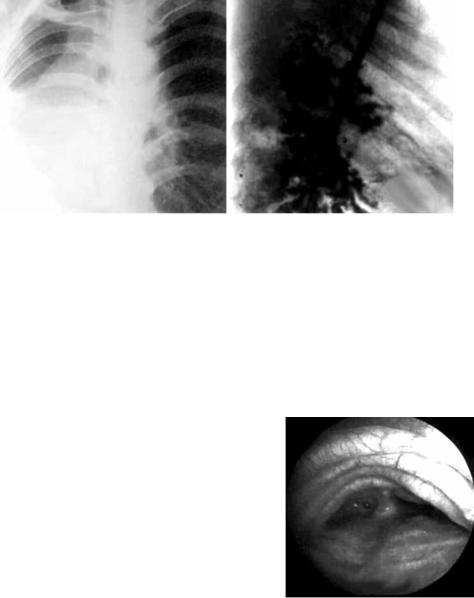
Рис. 8.4. Врождённая гипоплазия правого лёгкого (данные рентгенографии грудной клетки: слева – вид сбоку, справа – прямая проекция)
Приведено по: http://clinica.samsmu.ru/radio/radio.htm
2.Аномалии, обусловленные преимущественным нарушением развития бронхоэпителиального ветвления: трахеобронхомегалия и трахеобронхомаляция, стенозы трахеи и бронхов, лобарная эмфизема лёгких, дивертикулы трахеи и бронхов, бронхопищеводные свищи, бронхогенные кисты, врождённые бронхоэктазы, гамартохондромы.
3.Аномалии развития сосудов малого круга: аплазия сосудов, гипоплазия сосудов, аневризмы и артериовенозные свищи, аномалии впадения лёгочных вен.
4.Аномалии сосудов большого круга: лё-
гочная секвестрация (внелёгочная, внутрилёгочная), аномалии бронхиальных сосудов (аплазия, гипоплазия), доля непарной вены.
5. Аномалии других тканей и органов: дермоидные кисты, тератомы, муковисцедоз.
Из перечисленных форм пороков развития наиболее часто встречаются аномалии, обусловленные нарушением развития брон- хо-эпителиального ветвления: поликистоз – 39,4 %; бронхоэктазы – 24,5; бронхолёгочные кисты – 9,7 %.
Синдром Мунье–Куна. Врождённая тра-
хеобронхомегалия, нередко сочетающаяся с трахеобронхомаляцией (рис. 8.5).
Рис. 8.5. Синдром Мунье–Куна (данные трахеобронхоскопии)
Приведено по: http://med-ru.com/ content/view/3877/86/
299
8.3. Врождённые аномалии (пороки развития) челюстно-лицевой области
Классификация эпонимических симптомов и синдромов в соответствии с МКБ-10:
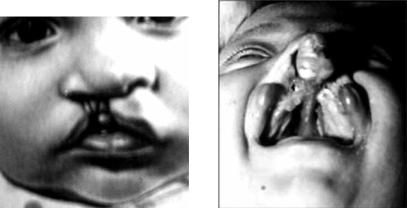
Q35 – расщелина нёба (волчья пасть);
Q36 – расщелина губы (заячья губа) (рис. 8.6);
Q37 – расщелина нёба и губы (волчья пасть с заячьей губой).
Отдельные эпонимические симптомы и синдромы.
Синдром Ван-дер-Вуда. Комплекс наследственных аномалий у человека (слизистые кистомы нижней губы, расщепление верхнего нёба и др.), передаваемый по аутосомно-доминантному типу (локус VWS расположен на участке q32–q41 хромосомы 1) (рис. 8.7).
Рис. 8.6. Расщелина верхней губы |
Рис. 8.7. Синдром Ван-дер-Вуда |
|
(заячья губа – labium leporinum) |
Приведено по: http://www.rusvrach.ru/ |
|
Приведено по: Н. Б. Ситковский |
||
img/data/medsestra-2– |
||
с соавт. (1981) |
2005str15ris6–7.jpg |
8.4. Врождённые аномалии (пороки развития) органов пищеварения
Классификация эпонимических симптомов и синдромов в соответствии с МКБ-10:
Q38 – другие врождённые аномалии (пороки развития) языка, рта и глотки; Q39 – врождённые аномалии (пороки развития) пищевода;
Q40 – другие врождённые аномалии (пороки развития) верхней части пищеварительного тракта;
Q41 – врождённые отсутствие, атрезия и стеноз тонкого кишечника; Q42 – врождённые отсутствие, атрезия и стеноз толстого кишечника; Q43 – другие врождённые аномалии (пороки развития) кишечника;
300