

Ч 2
.pdfОписан французскими дерматоло-
гами L. A. J. Brocа (1856–1928) и W. A. Dubreuilh (1857–1935).
Синдром Брюнауэра. Наслед-
ственная болезнь, характеризующаяся кератодермией (рис. 10.39), гипергидрозом и арковидным нёбом.
Описан австрийским дерматоло-
гом S. Brünauer.
Синдром Ваарденбурга. Наслед-
ственное заболевание, характеризующееся высокой изменчивостью симптомов, диагностическими из которых являются расширенные ноздри, гипертрихоз средней части бровей (в целом строение лица по типу «греческого профиля»), слабая пигментация кожи и волос, снижение остроты слуха и др. Наследуется по аутосомно-доминант-
ному типу (локус WS1 расположен на участке q34 хромосомы 9, имеются также данные о локализации этого локуса на участке q33–q37 хромосомы 2).
Описан в 1951 г. датским офтальмологом P. J. Waardenburg (1886–1979).
Синдром (болезнь) Бурневилля (синонимы – туберозный склероз, болезнь Бурневилля–Прингла, эпилойя). Наследственная болезнь из группы факоматозов, характеризующаяся сочетанием судорожных припадков, слабоумия и поражений кожи (фибромы, аденомы сальных желёз, пигментированные пятна кофейного цвета). Наследуется по аутосомно-доминантному типу.
Описан французским врачом D.-M. Boumeville (1840–1909) и английским врачом J. J. Pringle (1855–1922).
Синдром Вискотта–Олдрича. Наследственное заболевание человека, характеризующееся иммунодефицитом, экземой, редукцией размеров тромбоцитов (рис. 10.40). Локус WAS, расположенный на участке p11.22–p11.23 X-хромосомы, кодирует белок WASP с молекулярной массой 56 кДа, который выполняет адаптерные функции при внутриклеточной передаче сигнала во время агрегации тромбоцитов, индуцируемой тромбином, после их взаимодействия с волокнами коллагена. с-Концевая часть WASP взаимодействует с белковым комплексом
351

Arp2/3, участвующим в перестройках цитоскелета, а также с GTPазой
CDC42Hs-GTP, принадлежащей Rho-
семейству, с последующим их транспортом в подосомы. Через белок CrcL WASP взаимодействует с тирозиновой протеинкиназой syk – основным участником передачи сигналов во время активации тромбоцитов коллагеном. Известно более 100 мутаций в гене WAS, большинство из которых изменяет аминокислоты N-концевой части белка WASP.
Описан в 1949 г. немецким педиатром A. Wiskott и американким педиат-
ром R. Aldrich.
Синдром Габера. Наследуемая по аутосомно-доминантному типу болезнь, характеризующаяся сочетанием сыпи на лице, напоминающей розовые угри, с повышенной чувствительностью к солнечному свету и интраэпидермальными эпителиомами на коже туловища. Проявляется в детском возрасте.
Описан в 1971 г. английским дерматологом Н. Haber.
Синдром Гельмгольца–Харрингтона. Редкое заболевание, проявляю-
щееся врождённым помутнением роговицы, костными деформациями, гепато- и спленомегалией. Отмечают короткие толстые лапоподобные руки и ноги, поясничный кифоз, ограничение движений рук и ног, ладьеобразную голову и существенное отставание в росте. В ряде случаев выявляется гиперспленизм. Причина не известна.
Описан в 1911 г. американскими врачами H. F. Helmholz и E. R. Harrington.
Синдром Гертвига–Веперса. Наследственное заболевание, характеризующееся олигодактилией указательного и большого пальцев, отсутствием лучевой кости, остроугольным анкилозом локтевого сустава, недоразвитием и синостозом грудного сегмента, пороками развития почек.
Впервые описан немецким эмбриологом К. W. Th. R. Hertwig (1850–1937). Синдром Гольденхара. Сочетание аномалий развития зубов, ушной раковины и слухового прохода с гипоплазией половины лица и дермоидной кистой
или липодермоидом конъюнктивы (рис. 10.41).
Описан в 1966 г. американским врачом М. Goldenhar.
352

Рис. 10.41. Внешний вид пациента и изменения со стороны черепа при синдроме Гольденхара
Приведено по: http://www.senyumbali.org/newsletter/newsletter03/newsletter03_html_746c03a7. jpg, http://www.mypacs. net/repos/mpv3_repo/viz/full/17349/867477.jpg
Синдром Гольтца–Горлина (синоним – эктомезодермальная наслед-
ственно-семейная аномалия). Наследуемая по аутосомно-доминантному типу болезнь, проявляющаяся образованием резко отграниченных очагов истончённой гиперпигментированной кожи, дистрофией ногтей, гипотрихозом, аномалиями развития глаз, гортани, сердца, скелета и т. д.
Описан американскими врачами R. W. Goltz и R. J. Gorlin.
Синдром Граухана. Наследственный порок развития – многопалость, губно-нёбная щель, пороки развития почек, мочевого пузыря и половых органов (образование щелей).
Описан немецким хирургом М. Grauhan (1886–1945).
Синдром Грейга (синонимы – глазной наследственный гипертелоризм, семейный гипертелоризм, гипертелоризм Грейга). На-
следственная семейная аномалия развития, характеризующаяся широко расставленными глазами, широкой спинкой носа, микроцефалией
ималым ростом, иногда сочетающимися с крипторхизмом, грыжами, аномалиями челюстей
изубов,олигофренией, эпилепсией идр.(рис.10.42). Наследуется по аутосомно-доминантному типу.
Локус GCPS расположен на участке p13 хромо- |
Рис. 10.42. Внешний вид ребёнка |
|
сомы 7 (предполагается, что заболевание обу- |
||
словлено мутациями гена GLI3, кодирующего |
с синдромом Грейга |
|
Приведено по: http://upload. |
||
хроматин-ассоциированный фосфопротеин). |
||
wikimedia.org/wikipedia/commons/ |
||
Описан в 1924 г. английским врачом |
thumb/5/5a/Greig_syndrome. |
|
D. М. Greig (1864–1936). |
JPG/190px-Greig_syndrome. JPG |
353
Синдром Грефе–Шегрена. Наследственная болезнь, характеризующаяся олигофренией, шизофреноподобным синдромом, мозжечковой атаксией, глухотой или тугоухостью, пигментным ретинитом, катарактой, часто сочетающимися с маленьким ростом, микроцефалией, деформацией стоп и искривлением позвоночника. Наследуется по аутосомно-рецессивному типу.
Впервые был описан немецким врачом А. Graefe (1828–1870).
Синдром Груббера (синонимы – спланхнокистозная дизэнцефалия,
синдром Меккеля). Наследственная болезнь, характеризующаяся аномалиями развития черепа, позвоночника, мочеполовых органов, полидактилией, образованием кист в почках (реже – в печени, поджелудочной железе, яичниках). Наследуется по аутосомно-рецессивному типу.
Описан в 1923 г. немецким патологоанатомом G. В. Gruber.
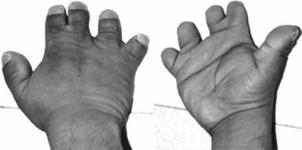
Синдром Де Ври. Наследственное сочетание геморрагического синдрома с аномалиями развития ладони и синдактилией II и III пальцев кисти
(рис. 10.43).
Описан в 1978 г. израильским педиатром А. De Vries.
Синдром Джакомини (синоним – истинная микроцефалия). Врождён-
ное заболевание, проявляющееся сочетанием микроцефалии, психического недоразвития, спастических параличей и судорог.
Описан итальянским анатомом С. Giacomini (1841–1898).
Синдром Джеруэлла–Ланге-Нилсена (синоним – Jerwell–Lange-Nielsen syndrome). Врождённый симптомокомплекс в виде сочетания тугоухости с периодически возникающими приступами фибрилляции предсердий и желудочков сердца.
Описан врачами A. Jerwell и F. Lange-Nielsen.
Синдром Демарке–Рише. Порок развития, наследуемый по доминантному типу, при котором наблюдаются заячья губа, волчья пасть, свищи нижней губы, низкий рост, инфантилизм и клинодактилия. В ряде случаев могут быть комбинированный порок сердца, сакрализация поясничных позвонков и сколиоз.
Синдром Де Санктиса (синонимы – ксеродермическая идиотия). На-
следственная болезнь, проявляющаяся слабоумием различной тяжести, пиг-
Рис. 10.43. Кисти рук при синдроме Де Ври
Приведено по: http://www.nature. com/ejhg/journal/v15/n7/images/5201833f1.jpg
354


Синдром Кронкайта–Канада. Редко встречающийся комплекс врождённых аномалий с генерализованным желудочно-кишечным полипозом (рис. 10.45) и кожными проявлениями. Наследуется по аутосомно-доминантному типу. Проявляется кистозным полипозом большой кривизны желудка, двенадцатиперстной и начальной части тощей кишки, сочетающимся с разнообразными кожными проявлениями и энтеропатическими потерями белка. Выявляются дистрофические изменения кожи, алопеция, атрофия (дистрофия) ногтей и кожная гиперпигментация. Характерны боли в животе, рвота желудочным содержимым с примесью крови, диарея (иногда кровянистого характера), потеря массы тела и гипопротеинемия. При длительном течении наблюдаются отёки, асцит, кишечные кровотечения и кахексия. Описаны варианты течения данного синдрома, когда наряду с вовлечением в процесс только желудка отмечается облысение. Морфологически чаще всего выявляют гипертрофию слизистой желудка и тонкой кишки (что расценивается как следствие гипертрофического гастрита и энтерита и отождествляется с аномальным полипозом пищеварительного тракта). Имеет место снижение в крови уровней кальция и магния, нарастает гипопротеинемия, обусловленная синдромом мальабсорбции и приводящая к тяжёлым (порой необратимым) изменениям в организме. Возможна малигнизация полипов пищеварительного тракта.
Описан в 1955 г. американскими врачами L. М. Cronkhite и W. J. Canada.
Синдром Корнелия де Ланге (синонимы – синдром Брахмана–де Ланге
II, болезнь Брука–де Ланге). Заболевание, проявляющееся карликовостью, срастанием бровей на переносице, гирсутизмом, низко посаженными ушами, широко расставленными зубами, обезьяноподобной одиночной пальмарной складкой, аномалиями развития костной системы (брахицефалией, гипертелоризмом, гипоплазией нижней челюсти, малым размером кистей и стоп и др.), мышечной гипертрофией, низким коэффициентом интеллекта и судо-
Рис. 10.45. Структура полипа кишечника при синдроме Кронкайта–Канада Приведено по: http://www.gastro.org/user-assets/Images/edigest_new/IOM/CGH0626.jpg
356

рожными припадками (рис. 10.46). Заболева- |
|
ние носит врождённый характер, однако хро- |
|
мосомной патологии установить не удалось. |
|
Описан голландским психиатром С. de Lan- |
|
ge (1871–1950). |
|
Синдром (болезнь) Лейна II (синонимы – |
|
симметричная невиформная акроэритема, |
|
наследственная эритема ладоней и подошв). |
|
Наследственная симметричная эритема ладо- |
|
ней (иногда и подошв), которая, как предпо- |
|
лагается, является разновидностью наслед- |
|
ственной кератодермии. Наследуется по ауто- |
|
сомно-доминантному типу. |
Рис. 10.46. Внешний вид ребёнка |
Описан в 1949 г. американским врачом |
|
J. Е. Lane. |
с синдромом Корнелия де Ланге |
Синдром Летюлля (синонимы – кисто- |
Приведено по: http://www.psychnet- |
видная лимфома, кистовидная гигрома). |
uk.com/dsm_iv/pictures/9.jpg |
|
Порок развития – ячеистая лимфангиома на шее, языке, губе у новорождённых.
Описан в 1882 г. врачом M. Letulle.
Синдром Лешке (синоним – наследственная пигментная дистрофия).
Наследственная болезнь, характеризующаяся нарушением деятельности вегетативной нервной системы, дисфункцией гипофиза и надпочечников, инфантилизмом и многочисленными пигментными пятнами на коже.
Описан немецким терапевтом Е. F. W. Leschke (1887–1933).
Синдром Лоренса–Муна–Барде–Бидля (синонимы – синдром Барде,
синдром Бидля). Наследственная болезнь, характеризующаяся ожирением, гипогенитализмом, пигментным ретинитом, задержкой умственного развития, полидактилией и синдактилией. Предположительно наследуется по ауто- сомно-рецессивному типу. Пациенты (мужского пола) страдают патологией зрения (ночная слепота, косоглазие, катаракта, другие нарушения вплоть до потери зрения), имеют маленькие гениталии, скудный рост волос на теле, азоспермию, сниженный коэффициент интеллекта, аномалии почек (вздутие чашечек, кисты или дивертикулы, фетальная дольчатость почек и терминальная стадия почечной недостаточности – до 15 %).
Описан английским офтальмологом J. Z. Laurence (1830–1874), американским офтальмологом R. Ch. Moon (1844–1914), французским врачом G. Bardet (1885–1932) и чешским врачом А. Biedl (1869–1933).
Синдром Лоу (синоним – окулоцереброренальный синдром). Наслед-
ственная болезнь, характеризующаяся поражением глаз (катаракта и глаукома) (рис. 10.47), умственной отсталостью, повышенным выделением с мочой органических кислот.
Описана в 1962 г. американским педиатром Ch. U. Lowe.
357

Синдром Маделунга (синонимы – диффузная липома шеи, доброкачественный симметричный липоматоз, жирная шея Маделунга, синдром Лонуа–Бансода). Разновидность наследственного множественного липоматоза. Тип наследования – аутосомнодоминантный. Жировые разрастания охватывают переднебоковые и задние поверхности шеи, распространяясь далее на подбородок и грудную клетку. Иногда отмечаются множественные симметричные липоматозные узлы на туловище и конечностях. Течение обычно медленное, но иногда наблюдается быстрый рост опухолей. Боль
возникает при значительном увеличении количества жировой ткани, когда сдавливаются периферические нервы, нарушается венозный отток. Могут сдавливаться гортань, глотка, что проявляется расстройством дыхания, дисфагией, дизартрией, стенокардией, псевдомиопатией, постепенным нарастанием слабости в мышцах конечностей. Болеют преимущественно мужчины в возрасте 35–40 лет. Лабораторные сдвиги не обнаруживаются. При гистологическом обследовании выявляют скопления жировой ткани нормальной структуры.
Описан немецким хирургом О. W. Madelung (1846–1926).
Синдром Мак-Кьюна–Олбрайта.
Врождённое заболевание, проявляющееся фиброзной остеодисплазией (рис. 10.48), гиперпигментацией кожных покровов, асимметрией лица и преждевременным половым созреванием. Рентгенограмма черепа показывает увеличение плотности костей основания черепа.
Впервые описан американским врачом F. Albright, американским педиатром D. J. McCune и американским патологом W. Н. Sternberg.
Синдром Мак-Кьюсика (синоним – метафизарная дисплазия).
Врождённое заболевание с аутосом- но-рецессивным типом наследования, проявляющееся карликовым ростом
358

с относительно длинным туловищем и короткими конечностями. Укороченные пальцы рук расположены наподобие спиц в колесе. Перерастяжимость суставов кистей и пальцев сочетается с тугоподвижностью локтевых суставов. Часто отмечается ломкость и укороченность ногтей. Грудная клетка нередко колоколообразной формы. Возможно наличие кишечных аномалий – мегаколона, мальабсорбции. У взрослых развивается ожирение. Рентгенологические изменения чаще обнаруживаются в коленных суставах и коротких трубчатых костях: метафизарные линии волнистой конфигурации, резко отграниченные зоны обызвествления расширены, утолщены, иногда с кистозными изменениями.
Описан врачом-генетиком V. A. McKusick.
Синдром Манкрифа. Триада наследственных аномалий – грыжа пищеводного отверстия диафрагмы, задержка умственного и физического развития, алиментарная дисахаридурия (сахарозурия, лактозурия и фруктозурия). Иногда сочетается с аномалиями развития головного мозга.
Синдром Марфана. Наследственная болезнь соединительной ткани (рис. 10.49). Заболевание наследуется по аутосомно-доминантному типу, обусловлено мутациями в гене фибриллина (гликопротеин с молекулярной массой 350 кД – основной компонент внеклеточных микрофибрилл), локализо-
Рис. 10.49. Внешний вид пациентки с синдромом Марфана Приведено по: http://www.critical.ru/actual/marfan/Ris1–2.htm
359
ванном на длинном плече хромосомы 15. Характеризуется вариабельностью фенотипических проявлений, включая сердечно-сосудистые, глазные, мы- шечно-скелетные признаки и поражение центральной нервной системы. Встречается в 1–3 случаях на 5000 человек, не имеет расовой и половой детерминированности. Относится к генным болезням с высокой пенетрантностью и различной степенью экспрессивности. В 70–85 % случаев заболевание является наследственным, в остальных – развивается вследствие спонтанных мутаций. Причиной синдрома Марфана в 95 % случаев являются мутации в гене фибриллина-1 и фибриллина-2 (локализация в хромосомах 15q21 и 3p24–p25). Фибриллин – гликопротеид, обеспечивающий основу эластическим волокнам соединительной ткани (он находится в межклеточном матриксе, хрящах, стенках сосудов, хрусталике глаз и др.). В 5 % случаев при синдроме Марфана описывают мутации в a2-цепи коллагена I типа. Синдром Марфана (Q87.4) в МКБ-10 рассматривается в Классе XVII «Врождённые аномалии (пороки развития), деформации и хромосомные наруше - ния»; он внесен в каталог V. A. McKusick «Менделевское наследование у человека: каталог человеческих генов и генетических болезней» (MIM), ему присвоен номер 154700.
Современные критерии для диагностики синдрома Марфана:
со стороны скелета – наличие не менее четырёх из следующих больших признаков либо наличие двух больших критериев или одного большого и двух малых:
большие признаки: воронкообразная или килевидная грудная клетка (нуждается в хирургическом лечении), уменьшение верхнего сегмента тела (расстояние от головы до лобка) по отношению к нижнему (расстояние от лобка до пола) или соотношение размаха рук к росту более 1,05, положительный тест запястья или большого пальца, сколиоз более 20º или спондилолистез, ограничение разгибания в локтевом суставе менее 170º, протрузия вертлужной впадины любой степени (устанавливается при рентгенологическом исследовании), медиальное смещение внутренних лодыжек вследствие продольного плоскостопия;
малые признаки: мягкая экскавация грудины, гипермобильность суставов, арковидное нёбо и скученность зубов, характерное лицо;
со стороны органов зрения – наличие одного большого (эктопия хрусталика) или двух малых признаков – уплощение роговицы (устанавливается при кератометрии), увеличение аксиального размера глазного яблока (устанавливается при ультрасонографии), гипоплазия радужной оболочки или цилиарной мышцы;
со стороны сердечнососудистой системы – наличие одного большого признака (дилатация корня аорты с/без регургитации и с вовлечением синусов Вальсальвы, расслоение восходящей аорты) или одного малого (аортальная недостаточность, пролапс митрального клапана с/без митральной регургитации, дилатация ствола лёгочной артерии при отсутствии клапанного либо
360