

Ч 2
.pdfСиндром Каннера (синоним – детский аутизм). Наследственное заболе-
вание человека, проявляющееся аутизмом (погружением в мир собственных переживаний), который является ранним проявлением шизофрении и характеризуется отсутствием способности решать элементарные вопросы (может сочетаться с исключительными способностями в других областях). Предполагается, что болезнь обусловлена дефицитом аденилсукциназы (ген ADSL локализован на участке q12–qter хромосомы 22).
Описан в 1936 г. австрийским психиатром L. Kanner.
Синдром Карпентера (синонимы – аутоиммунный полигландулярный синдром II типа, акроцефалополисиндактилия II типа). Аутоиммунный полигландулярный синдром (АПГС), представляющий собой первичное поражение эндокринных желёз с формированием их функциональной недостаточности. Часто сочетается с различными органоспецифическими неэндокринными заболеваниями аутоиммунного генеза. АПГС II типа – наиболее распространённый вариант иммуноэндокринных нарушений. Характеризуется поражением эндокринных желёз с развитием первичного гипотиреоза, сахарного диабета I типа, первичного гипогонадизма, миастении и стеатореи. Этим проявлениям нередко сопутствуют витилиго, деформации лицевого черепа, аллопеция и пернициозная анемия (рис. 7.30).
Все перечисленные заболевания, встречающиеся в комбинации АПГС II типа, ассоциированы главным образом с HLA (с повышенной частотой отмечаются гаплотипы HLA B8, Dw3, Dr3, Dr4). В большинстве случаев синдром проявляется спорадически, при семейных формах в различных вариантах может отмечаться в нескольких поколениях.
Описан английским педиатром G. Carpenter (1859–1910).
Синдром Коккейна. Наследственная болезнь, характеризующаяся нарушением обмена веществ и проявляющаяся карликовостью в сочетании с микроцефалией и кахексией (рис. 7.31). Наследуется по аутосомно-рецессивному типу.
Впервые описан английским педиатром E. A. Cockayne (1880–1956). Синдром Кокрена. Наследственное заболевание, характеризующееся ано-
малией метаболизма. Проявляется гипогликемией, провоцируемой введением белковой пищи (лейцином). Проявляется со дня рождения в виде отказа от пищи, сильной потливости, рвоты, общего беспокойства ребёнка. Ухудшение состояния отмечается после приёма молока, яиц, мяса, рыбы. При лабораторном исследовании крови наблюдается нормальный уровень глюкозы крови, однако после нагрузки лейцином регистрируются гипогликемия, гиперинсулинемия. Прогноз неблагоприятный – гипогликемия сопровождается клони- ко-тоническими судорогами, часто заканчивающимися смертельным исходом. Ограничение ребёнка в еде ведёт к нарушениям, типичным для белкового голодания.
Описан английским педиатром W. А. Cochrane.
251

Рис. 7.30. Проявления синдрома Карпентера: а – внешний вид пациента; б – изменения ко - нечностей; в – характерные изменения со стороны черепа; г – рентгенологические изменения конечностей
Приведено по: http://download.ajhg.org/images/journalimages/0002–9297/ PIIS0002929707610337. gr1_lrg.hi.jpg
252

Синдром Криглера–Найяра. Врождён-
ная негемолитическая желтуха, обусловленная нарушением обмена билирубина у детей вследствие недостатка фермента УДФ-глю- куронозилтрансферазы. Передаётся по ауто- сомно-рецессивному типу (ген UDPGT локализован на длинном плече хромосомы 2).
Описан в 1951 г. американскими педиатра-
ми J. F. Crigler и V. A. Najjar.
Синдром Кэнни–Каффи. Врождённое за-
болевание с аутосомно-доминантным типом наследования, проявляющееся низким ростом, остеосклерозом, кортикальным утолщением длинных трубчатых костей, поздним закрытием передних каналов fontanel, кальцификацией базальных ганглиев, анофтальмией или гиперметропией. Одним из главных проявлений болезни является выраженный гипопаратиреоз (ген, ответственный за развитие синдрома, локализован на 1q42–43). Молеку- лярно-генетические исследования идентифицировали мутации гена ТВСЕ, кодирующего тубулин-специфические шаперон-белки, которые необходимы для приобретения правильной конформации (фолдинга) а-субъ- единиц тубулина и образования а-b-тубулин- гетеродимеров. При тщательной ревизии на аутопсии в случаях гибели пациентов с данным синдромом при наличии признаков гипопаратиреоза паращитовидные железы вообще
не были обнаружены, что предполагает эмбриональную причину их отсутствия.
Синдром Лайтвуда. Вариант наследственного синдрома Лайтвуда–Ол- брайта, обусловленного неспособностью проксимальных извитых почечных канальцев реабсорбировать анионы бикарбоната. Наблюдается в детском возрасте.
Описан в 1939 г. английским педиатром R. Lightwood.
Синдром Лайтвуда–Олбрайта (синоним – семейный почечный ацидоз).
Наследственное сочетание полиурии, рахитоподобных изменений скелета, нефрокальциноза, нефролитиаза и нарушений кислотно-щелочного равновесия. Различают синдром Лайтвуда и синдром Олбрайта–Баттлера.
Описан в 1938 г. английским педиатром R. Lightwood и американским вра-
чом F. Albright.
253

Синдром (болезнь) Лаки–Лоран-
да(синоним–Laki–Lorandsyndrome).
Наследственный геморрагический диатез, обусловленный нарушением образования фибрина в связи с дефицитом XIII фактора свёртывания крови. Наследуется по аутосомно-рецессив- ному типу.
Описан врачами К. Laki и L. Lorand.
Синдром Ландштейнера–Фанко- ни–Андерсена (синонимы – муковисцидоз, кистозный фиброз поджелудочной железы, триада Андерсена,
кистозный фиброз поджелудочной железы, врождённая семейная бронхопанкреатическая дистрофия). На-
следственное заболевание с аутосом- но-рецессивным типом наследования, проявляющееся тяжёлыми расстройствами функции органов дыхания (рис. 7.32), желудочно-кишечного трак-
та и ряда других органов и систем с множественными поражениями желёз внутренней секреции. Изменённый в качественном и количественном составе секрет поджелудочной железы, желудочных и кишечных желёз, бронхов, гепатобилиарной системы, слюнных желёз застаивается в выводных протоках. Закупорка их вязким секретом приводит к повышенному давлению в протоковой системе, её кистозной трансформации и гибели железистого аппарата органа, после чего секреция прекращается.
Выделяют следующие клинические формы заболевания: смешанную (лё- гочно-кишечную), преимущественно лёгочную (рис. 7.33), преимущественно кишечную, мекониальный илеус, абортивные и стёртые формы.
Кишечные проявления муковисцедоза проявляются с раннего детства и связаны с нарушением активности ферментов поджелудочной железы и кишечника. Клинически при этом отмечаются массивная стеаторея, креаторея, диарея (кал объёмный, бесцветный, блестящий, с гнилостным запахом). Гнилостные процессы в кишечнике способствуют вздутию живота и диарее. У некоторых пациентов отмечается выпадение прямой кишки и развитие цирроза печени.
Мекониальный илеус отмечается у 10–20 % новорождённых, страдающих муковисцидозом. Для него характерна клиника кишечной непроходимости с рвотой желчью, неотхождением мекония, увеличением живота, развитием перитонита. Ввиду дефицита жирорастворимых витаминов А, Е и К прогресси-
254

руют общая слабость, гиперкератоз, расстройства зрения, снижается устойчивость к инфекциям.
Лёгочные проявления муковисцидоза развиваются на 1–3-м году жизни и характеризуются рецидивирующими пневмониями, бронхитами с пневмосклеротическими изменениями, развитием эмфиземы лёгких и бронхоэктазов. Отмечаются надрывный кашель, одышка, цианоз.
Для подтверждения диагноза проводится исследование пота (ногтей, слюны) на содержание натрия и хлора, оценивается копрограмма. Прогноз неблагоприятный. Гибель пациентов происходит от септических осложнений (преимущественно со стороны дыхательной системы), печёночной и почечной недостаточности.
Впервые описан швейцарским пе-
диатром G. Fanconi (1892–1958) и аме-
риканским патологоанатомом D. Н. Andersen (1901–1964).
Синдром Лепе (синоним – наследственный щавелевокислый диатез). На-
следственное заболевание, характеризующееся хронической диспепсией, непереносимостью некоторых продуктов (шпинат, рис), недостаточностью функции поджелудочной железы и другими симптомами. Передается по аутосомно-ре- цессивному типу, обусловлен дефицитом фермента глиоксалатредуктазы.
Синдром Леша–Найхана. Наследственное заболевание человека, сопровождающееся неврологическими и психическими расстройствами и гиперуринемией, обусловленными врождённым дефицитом фермента гипоксантин-гуа- нин-фосфорибозилтрансферазы. Передается по рецессивному, сцепленному с полом, типу (ген HPRT локализован на участке q26 Х-хромосомы). Проявляется умственной отсталостью, хореоатетозом, приступами агрессивного поведения с самоповреждением (царапаньем лица и тела), повышенным содержанием мочевой кислоты в моче. У мальчиков уже в самом начале жизни заболевание проявляется спазмом сгибателей туловища, клоническими движениями, атетозом, мышечной слабостью, гипотонией и судорожными припадками.
Описан в 1980 г. американским педиатром М. Lesch и американским вра-
чом М. L. Nyhan.
Синдром Линча–Каплана–Хенна–Краша. Наследственная полиэндо-
кринопатия, проявляющаяся сахарным диабетом, гиперлипидемией, гипогонадизмом и низким ростом.
255

Синдром Луи-Бара (синоним – атаксия-телеангиэктазия). Наследствен-
ная болезнь из группы факоматозов, характеризующаяся медленно развивающимися мозжечковыми расстройствами, симметричными телеангиэктазиями и гемангиомами (рис. 7.34), особенно на конъюнктивах, коже лица и шеи, рецидивирующими пневмониями с развитием бронхоэктазов. Часто сочетается с гипоплазией вилочковой железы и дисгаммаглобулинемией. Проявляется в детском возрасте. Наследуется по аутосомно-рецессивному типу.
Описан в 1968 г. французским врачом D. Louis-Bar.
Синдром Мак-Куарри. Наследственная гипогликемия. Признаки заболевания выявляются до 3 лет жизни. Проявляется утром натощак приступами раздражительности, головокружения, потливости и мышечной слабости. Изредка бывают судороги. Лабораторное исследование показывает низкий уровень глюкозы в крови. При частом повторении приступов развивается атаксия, наблюдается задержка умственного развития. Этиология до настоящего времени не установлена.
Описан в 1934 г. американским педиатром I. Mac-Quarrie.
Синдром Мак-Леода. Редкое врождённое заболевание, характеризующееся акантоцитозом (своеобразная форма эритроцитов) и нарушением экспрессии антигенов групп крови Келла. Передаётся по рецессивному, сцепленному с полом, типу (локус ХК расположен на участке p21 Х-хромосомы).
Синдром Марото–Лами (синоним – мукополисахаридоз VI типа). На-
следственное заболевание человека, связанное с нарушением обмена мукополисахаридов – первичной недостаточностью арилсульфатазы В (N-ацетилгек- созамин-4-сульфатсульфатазы). Наследуется по аутосомно-рецессивному типу. Проявляется выраженными костными изменениями, помутнением роговицы, карликовым ростом, гротескными чертами лица, гепатоспленомегалией, контрактурой крупных и мелких суставов (рис. 7.35). Имеют место гидроцефалия, паралитическое сходящееся косоглазие, равномерное повышение сухожильных рефлексов. Проявления болезни отмечаются с раннего детского
Рис. 7.34. Симметричные гемангиомы на конечностях при синдроме Луи-Бара Приведено по: http:// 208.96.47.3/images/community/dermatlas/at_1_010907_thumb.png
256

Рис. 7.35. Контрактура суставов верхних и нижних конечностей и рентгенологические признаки искривления таза у девочки 9 лет с синдромом Марото–Лами
Приведено по: http://www.nedug.ru/library/doc.aspx?item=55684
возраста, но интеллект, как правило, не страдает. Отмечаются также гепатоспленомегалия, пупочная грыжа, в биохимических анализах крови определяется снижение активности аринсульфатазы, выявляется повышенная экскреция с мочой дерматансульфата. При рентгенографии отмечается увеличение размеров черепа, недостаточная сомкнутость черепных швов, углубление турецкого седла с расширением входа в него, уплощение рёбер и позвонков, неравномерность длины и формы костей пястны, плюсны и фаланг пальцев. При люмбальной пункции определяется высокое давление спинно-мозговой жидкости (до 300 мм вод. ст.), белок– 1,6 мг/л, цитоз – 2 лимфоцита в 1 мкл.
Описан в 1963 г. P. Maroteaux и M. Lamy.
Синдром Мартина–Олбрайта (синоним – псевдогипопаратиреоидный синдром Мартина–Олбрайта). Симптомокомплекс, обусловленный врождённым нарушением утилизации тканями гормонов паращитовидных желёз. Характеризуется гипокальциемией, гиперфосфатемией, ахондроплазией, а также неполноценностью умственного развития, частыми приступами тетании, развитием катаракты, дистрофией ногтей и другими признаками.
Описан в 1942 г. швейцарскими врачами Е. Martin и F. Albright.
Синдром Менкеса (синонимы – copper transport disease, болезнь «кур-
чавых волос»). Наследственное заболевание, обусловленное врождённым нарушением всасывания меди в кишечнике (гипокупремия). Наследуется по рецессивному, сцепленному с полом, типу (локус MNK расположен на участке
257
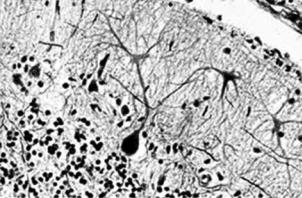
Рис. 7.36. Дегенеративные изменения в головном мозге при синдроме Менкеса Приведено по: http://www.ncbi.nlm.nih.gov/disease/GIFS/ATP7A.gif
q13.2–q13.3 Х-хромосомы). Активность ряда ферментов (супероксиддисмутазы, тирозиназы и др.) при этом снижена, отмечаются тяжёлая дегенерация нервной системы (рис. 7.36), истончение волос и образование на них характерных вздутий. Обычно в возрасте 1–2 лет наступает летальный исход.
Описан в 1962 г. американским педиатром J. Н. Menkes.
Синдром Монкриффа. Врождённое нарушение углеводного обмена, сопровождающееся лакто- и фруктозурией, алиментарной сахароурией. Проявляется задержкой общего развития (физического и психического) и наличием диафрагмальной грыжи.
Описан в 1969 г. английским педиатром А. Moncrieff.
Синдром (болезнь) Моркио (синоним – мукополисахаридоз IV типа).
Врождённое заболевание из группы мукополисахаридозов (1:40 000 новорождённых), характеризующееся множественными поражениями скелета из-за нарушения катаболизма мукополисахаридов (кислых гликозаминогликанов) в результате дефицита хондроитин-N-ацетилгексозоамино-6-сульфатсульфа- тазы (тип А, ген этого фермента локализован на участке q24.3 хромосомы 16 человека) или D-галактозидазы (тип В). Наследуется по аутосомно-рецессив- ному типу.
Характеризуется нормальным психическим развитием и характерной дистрофией костей, которую можно классифицировать как спондилоэпифизарную дисплазию (рис. 7.37). Резко выраженная гипоплазия зубовидного отростка может вызывать кривошею и приводит обычно к той или иной степени компрессии спинного мозга. Довольно часто выявляют недостаточность аортального клапана с регургитацией крови. У пациента могут отмечаться карликовость, глухота и мышечная слабость. Обращают на себя внимание большой, грубо очерченный рот, широко поставленные зубы, «куриная», килевидная или бочкообразная грудная клетка. Отмечается кифоз или сколиоз грудного и поясничного отделов позвоночника. Питание снижено. Возникают контрак-
258

Рис. 7.37. Типичный внешний вид и рентгенологические изменения со стороны позвоночника (искривление, платиспондилия) при синдроме Моркио
Приведено по: http://www.nedug.ru/library/doc.aspx?item=55684
туры в локтевых, плечевых и коленных суставах, отмечаются вальгусная деформация нижних конечностей и плоскостопие; мышечная сила снижена. В случае компрессии спинного мозга помимо мышечной гипотонии отмечается поражение пирамидной системы, возможно развитие параплегии, паралича дыхания. Кожа утолщена, её тургор и эластичность снижены. Часто выявляются пупочные и паховые грыжи, расхождение прямых мышц живота; нередко отмечаются снижение слуха, дистрофические процессы в роговице. Почти у всех пациентов, доживших до 20 лет, развивается глухота. Интеллект не снижен. В большинстве случаев летальный исход наступает до 20 лет вследствие сердечно-лёгочной недостаточности, развивающейся на фоне интеркуррентных заболеваний. Возможна внезапная смерть в результате смещения ат- ланто-окципитального сочленения и повреждения ствола мозга.
Описан в 1929 г. уругвайским педиатром L. Morquio (1867–1935) и англий-
ским врачом J. F. Braiisford.
Синдром (болезнь) Минковского–Шоффара–Генсслена (синонимы – синдром Генсслена–Эрба, семейная гемолитическая желтуха, наследственный сфероцитоз, сфероклеточная анемия, конституциональная гемолитическая желтуха, сфероклеточная болезнь, сфероцитарная наследствен-
ная анемия). Заболевание с аутосомно-доминантным типом наследования. Клинически проявляется в возрасте 3–10 лет, тяжесть клинических проявлений весьма вариабельна. Желтуха обычно непостоянная, анемия умеренная, вызывающая лишь небольшую бледность кожных покровов. Наблюдается спленомегалия (селезёнка плотная, безболезненная при пальпации). Иногда выявляются аномалии пигментации.
259
Втечении заболевания выделяют: 1) гемолитический криз (проявляется нарастанием анемии, сопровождающейся одышкой, тошнотой, рвотой, болями
вживоте, адинамией, повышением температуры тела до 38–40°С, увеличением печени и селезёнки), при этом в крови отмечается нормохромная анемия (доля ретикулоцитов составляет до 50–60 % и более); 2) апластический криз (характерны симптомы тяжёлой анемии без признаков желтухи, небольшие размеры селезёнки, гипохромная анемия, отсутствие ретикулярной реакции).
Вряде случаев при данном синдроме отмечаются башнеобразный или большой круглый череп, малый рост, аномалии развития зубов, глаз и ушей, врождённые пороки сердца, высокое нёбо, западение переносицы, кривошея, полидактилия и синдактилия. Во всех случаях заболевания при анализе крови определяются анемия, равное количество сфероцитов, снижение минимальной осмолярной стойкости эритроцитов и уменьшение их среднего размера, возможно появление нормобластов.
Описан в 1900 г. польским врачом О. Mińkowski (1858–1931) и француз-
ским врачом А. М. Е. Chauffard (1855–1932).
Синдром Незелофа (синонимы – алимфоцитоз, лимфоцитарная аплазия, лимфоцитарная дисгенезия, французский тип иммунопареза, Nezelof
syndrome). Наследственная болезнь, характеризующаяся отсутствием реакций клеточного иммунитета вследствие количественной недостаточности и качественной неполноценности лимфоцитов (при нормальном содержании иммуноглобулинов в плазме крови). Наследуется по аутосомно-рецессивному типу.
Описан врачом С. Nezelof.
Синдром (болезнь) Ниманна–Пика (синонимы – липоидный гистиоцитоз, фосфатидный липоидоз, ретикулёз нелейкемический, ретикулоэндотелиоз метаболический, липоидно-клеточная спленогепатомегалия, сфин-
гомиелиноз). Наследственный липоидоз с неуклонно прогрессирующим течением, обусловленный нарушением обмена сложных липидов с накоплением сфингомиелина в клетках ретикулоэндотелиальной и центральной нервной систем (что придаёт клеткам характерный пенистый вид – клетки Пика). Наследуется по аутосомно-pецессивному типу (ген SMPD1 локализован на участке p15 хромосомы 11). Проявляется сразу после рождения. Дети становятся малоподвижными, вялыми, апатичными, отказываются от груди. Периодически появляется рвота, отмечается отставание в физическом и психическом развитии, может быть умеренная гидроцефалия. Периоды апатии сменяются беспокойством, гипертермическими кризами и приступами асфиксии. Наблюдаются прогрессивное похудание ребёнка, связанное с потерей аппетита, увеличение размеров живота, гепато- и спленомегалия, асцит.
Клинически различают три типа болезни:
тип А (более 1/2 всех наблюдений) – проявляется с первых месяцев жизни выраженной гепато- и спленомегалией, задержкой физического и умственного развития, тяжёлыми неврологическими расстройствами; большинство детей погибает до 3 лет;
260