

Ч 2
.pdfВ основу современной классификации ПИД положено преимущественное поражение того или иного звена иммунитета. Согласно этой классификации, первичные иммунодефициты делят на 5 групп: 1) недостаточность гуморального иммунитета; 2) недостаточность клеточного иммунитета; 3) комбинированная недостаточность гуморального и клеточного иммунитета; 4) недостаточность фагоцитов; 5) недостаточность системы комплемента.
Недостаточность гуморального иммунитета составляет 50–60 % от всех ПИД и проявляется нарушением продукции антител. В эту группу входят изолированный дефицит IgA (распространенность – 1:500), изолированный дефицит иммуноглобулинов других классов, дефицит иммуноглобулинов нескольких классов. Возможна недостаточность гуморального иммунитета при нормальной концентрации иммуноглобулинов, что обусловлено снижением уровня антител к определённой группе антигенов (например, снижением уровня антител к углеводным антигенам бактериальной стенки).
Наиболее часто встречается изолированный дефицит IgA, который возникает в результате дефекта конечной дифференцировки плазматических клеток, секретирующих IgA. У большинства пациентов дефицит IgA протекает асимптоматично, и лишь у небольшого числа пациентов имеется предрасположенность к возникновению лёгочных и кишечных инфекций.
Недостаточность клеточного иммунитета составляет 5–10 % от всех первичных иммунодефицитов и проявляется нарушением пролиферации и дифференцировки Т-лимфоцитов или недостаточностью стволовых клеток. Первичное нарушение клеточного иммунитета в большинстве случаев сопровождается вторичным нарушением синтеза антител.
Несостоятельность Т-клеток обусловливает повышенную чувствительность организма к вирусам и грибам, подавление которых основано на реак-
ции клеточного иммунитета (синдром Ди Джорджи и синдром Незелофа,
обусловленные недоразвитием тимуса во время эмбриогенеза). У таких пациентов не выявляются реакции клеточного иммунитета, и хотя в их организме синтезируются антитела, гуморальный ответ ослаблен. Реакции клеточного иммунитета нарушены у пациентов и с синдромом Вискотта–Олдрича (экзема, тромбоцитопения, подверженность инфекциям), и с синдромом Луи– Бара (атаксия-телеангиоэктазия). Встречаются случаи недостаточности T-клеток, при которых в сыворотке крови пациентов содержатся цитотоксические антитела, избирательно поражающие T-лимфоциты.
Недостаточность В-лимфоцитов ведёт к врождённой агаммаглобулинемии Брутона (встречается только у мужчин). Гипогаммаглобулинемия проявляется также гнойными инфекциями. Недостаточность иммуноглобулинов обнаруживается у сильно недоношенных детей, когда падает уровень полученных от матери IgA. Комбинированная недостаточность гуморального и клеточного иммунитета составляет 20–25 % от всех первичных иммунодефицитов. В эту группу входят заболевания, обусловленные первичным нарушением пролиферации и дифференцировки В- и Т-лимфоцитов. Уменьшение
141
числа Т-лимфоцитов и уровня иммуноглобулинов в крови наиболее выражено при тяжёлом комбинированном иммунодефиците, который является одной из наиболее неблагоприятных форм врождённого иммунодефицита. Он характеризуется возникновением разнообразных инфекционных (вирусных, грибковых, бактериальных) заболеваний сразу после рождения, часто приводит к ранней (обычно на первом году жизни) гибели.
Комбинированной недостаточности гуморального и клеточного имму-
нитета часто сопутствуют другие врождённые заболевания (например, тромбоцитопения при синдроме Вискотта–Олдрича или врождённые пороки сердца и гипокальциемия при синдроме Ди Джорджи).
Недостаточность фагоцитов составляет 10–15 % от всех первичных иммунодефицитов. Она обусловлена нарушением пролиферации, дифференцировки, хемотаксиса нейтрофилов и макрофагов и собственно нарушением процесса фагоцитоза. Фагоцитарные клетки – полиморфноядерные лейкоциты и клетки моноцитарно-макрофагального ряда. Они выполняют важную роль в защите против бактериальных инфекций и других внутриклеточных микроорганизмов. Выраженная недостаточность полиморфноядерных лейко-
цитов (циклическая нейтропения, или синдром Костманна) может приве-
сти к развитию генерализованной бактериальной инфекции. Важное значение имеют два генетических дефекта, нарушающих функцию фагоцитов, с которыми связано возникновение тяжелых заболеваний (нередко с летальным исходом) – хронического гранулематоза (причиной которого является нарушение механизма восстановления кислорода) и недостаточности адгезии лейкоцитов, или LAD-синдрома (вследствие наличия дефектов в генах интегринов). Значительно реже из этой серии врождённых иммунопатий встречаются син-
дром Чедиака–Хигаши и синдром Гринелли.
Недостаточность системы комплемента составляет не более 2 % от всех ПИД. Она проявляется нарушением опсонизации, фагоцитоза и разрушения микроорганизмов, сопровождаясь тяжёлыми инфекциями, вплоть до развития сепсиса. Недостаточность комплемента часто наблюдается при аутоиммунных заболеваниях (например, при системной красной волчанке).
Дефицит компонентов классического пути активации комплемента (C1q, C1r и C1s, C4 или C2) вызывает предрасположенность к заболеваниям, обусловленным нарушениями в формировании и клиренсе иммунных комплексов (например, к развитию системной красной волчанки). Дефицит С3, фактора В или фактора 1 приводит к повышению чувствительности организма к гнойным инфекциям. Недостаточность терминальных компонентов С5–С8, а также компонентов альтернативного пути (фактора D и пропердина) создаёт особую предрасположенность к инфекциям, вызываемыми двумя видами
Neisseria – N. gonorrhoeae и N. meningitidis. Всё это указывает на важную роль альтернативного пути активации комплемента и лизирующего мембрану комплекса в элиминации бактерий указанных видов. Самые тяжёлые клинические проявления имеет нарушение функций комплемента, связанное с недо-
142
статочностью ингибитора С1, что приводит к наследственному ангионевротическому отёку. Заболевание передается как аутосомно-доминантный признак. Ингибитор C1 блокирует не только классический путь активации комплемента, но подавляет также активность связанных с ним элементов кининовой и плазминовой систем, а также системы свёртывания крови. Формы недостаточности компонентов комплемента наследуются преимущественно как ауто- сомно-рецессивные признаки.
Вторичный иммунодефицит (ВИД) – патологический синдром, манифестирующим признаком которого является ослабление функциональной активности иммунной системы, развивающееся вторично на фоне (вследствие) патологических процессов (заболеваний) и проявляющееся комплексом специфических морфологических, функциональных и клинических признаков, характеризующих несостоятельность клеточных, гуморальных и неспецифических звеньев противомикробной защиты организма.
В это понятие включены собственно вторичные (приобретённые) иммунодефициты, а также состояния, когда сама иммунная система становится мишенью для инфекционного агента (СПИД, Т-клеточный лейкоз и др.). ВИД развивается гораздо чаще, чем ПИД, и формируется у лиц с исходно нормальной функцией иммунной системы. При вторичной иммунологической недостаточности могут поражаться Т-, В-системы иммунитета, а также факторы естественной резистентности (фагоцитоз, комплемент, интерфероны и др.), вероятно сочетанное их поражение. Это приводит к снижению защитных функций иммунной системы, нарушению регуляторных взаимоотношений между её компонентами.
Причины развития ВИД многообразны. Чаще всего ими являются: вирусные инфекции (ВИЧ-инфекция, грипп, эпидемический паротит, ве-
тряная оспа, корь, краснуха, острые и хронические гепатиты и др.); бактериальные инфекции (стафилококковые, стрептококковые, менинго-
кокковые, пневмококковые инфекции, сифилис, туберкулёз и др.); глистные и протозойные болезни (лейшманиоз, малярия, трихинеллёз,
токсоплазмоз и др.); злокачественные новообразования;
хронические, длительно текущие заболевания инфекционной и неинфекционной природы (хронические заболевания лёгких, мочевыводящей системы, сердечно-сосудистой системы, желудочно-кишечного тракта, системные заболевания соединительной ткани, дисбактериозы и др.);
нарушения питания (истощение, ожирение, микроэлементная и белковая недостаточность, авитаминоз, гиповитаминоз, нарушения всасывания и расщепления питательных веществ, длительное соблюдение строгих диет, несбалансированность питания по количественным и качественным составляющим и др.); влияние химиопрепаратов, а также средств, оказывающих иммунодепрессивное воздействие (цитостатиков, стероидных гормонов, антибиотиков, ни-
трофуранов и т. д.);
143
действие ионизирующей радиации и иммунотоксинов (в том числе ксенобиотиков), продолжительные стрессорные воздействия, переутомление;
патология обмена веществ (сахарный диабет, дефицит микроэлементов, гипербилирубинемия, недостаточность карбоксилазы и др.);
эндокринные нарушения (заболевания щитовидной железы, надпочечников; заболевания, связанные с нарушением центральных механизмов регуляции эндокринных функций и др.);
травмы, операции, ожоги и др.; возраст (у детей раннего возраста – в связи с незрелостью иммунной си-
стемы; у лиц пожилого возраста – в связи с угнетением клеточных иммунных реакций, падением активности антител и др.).
Таким образом, ВИД может возникать при действии на организм огромного числа социальных, экологических, медицинских, профессиональных и других факторов. Доля ВИД среди популяции людей достигает в отдельных сообществах 80–90 %.
Клинические проявления иммунных нарушений при ВИД могут быть: компенсированными (для этой формы ВИД характерна повышенная восприимчивость к возбудителям инфекций, что выражается в виде частых ОРВИ, пневмоний, пиодермий и т. д.), субкомпенсированными (для этой формы характерна склонность к хронизации инфекционных процессов, что клинически выражается в развитии хронических бронхитов, пневмоний, пиелонефритов, дуоденитов, панкреатитов, холециститов и др.), декомпенсированными, проявляющимися в виде развития генерализованных инфекций, этиологическим фактором развития которых является условно-патогенная микрофлора, и злокачественных новообразований (ярким примером декомпенсированной формы ВИД является СПИД).
Клинические проявления ВИД чрезвычайно разнообразны и характеризуются четырьмя основными синдромами:
инфекционным – проявляется рецидивирующим характером течения острых и хронических инфекционно-воспалительных заболеваний различной этиологии и локализации, гнойно-воспалительными инфекциями, вызываемыми условно-патогенными микробами;
аллергическим – проявляется аллергическими реакциями и заболеваниями; аутоиммунным – проявляется самостоятельными аутоиммунными нозологическими формами либо аутоиммунным компонентом на фоне длительного течения патологического процесса (поражение внутренних органов и систем); иммунопролиферативным – проявляется развитием опухолевого процес-
са, т. е. злокачественных опухолей, в различных органах и системах. Учитывая многообразие и широкую распространённость факторов, кото-
рые потенциально могут привести к развитию вторичной иммунологической недостаточности, логично предположить, что каждый человек в течение своей жизни подвергается длительному воздействию тех или иных факторов или их сочетаний, а следовательно, реальному риску развития ВИД.
144
С эпонимической точки зрения целесообразно выделить следующие про - явления первичного и вторичного иммунодефицита, различных иммунопатий у хирургических пациентов.
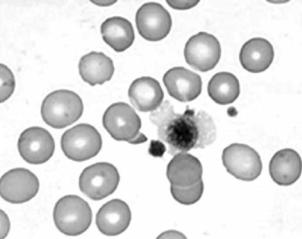
Аномалия Мей–Хегглина. Редкое врождённое заболевание с аутосомнодоминантным типом наследования, которое проявляется наличием гигантских тромбоцитов и больших базофильных включений в гранулоцитах (рис. 4.16). У некоторых пациентов наблюдается склонность к кровотечению, развивается тромбоцитопения с количественными или качественными аномалиями тромбоцитов, следствием чего является развитие тяжёлых гнойных рецидивирующих инфекций.
Аномалия Пельгера–Хюэта. Врождённое нарушение сегментации ядер гранулоцитов с аутосомно-доминантным типом наследования. При этом зрелые гранулоциты гетерозигот имеют только две доли в ядре и называются пенснеобразными клетками, поскольку напоминают по внешнему виду пенсне. Ядро гомозигот состоит только из одной доли. Клинически данная аномалия проявляется иммунодефицитом, сопровождаемым частыми инфекционными заболеваниями.
Впервые данная аномалия описана голландским врачом К. Pelger (1885– 1931), в широкую медицинскую практику термин введён голландским врачом
G. Т. Huët.
Болезнь Альдера–Рейли. Врождённая аномалия, при которой не нарушается функция нейтрофилов. При этом за счёт изменения метаболизма полисахаридов появляются окрашенные в сиреневый цвет азурофильные гранулы. Заболевание протекает с явлениями иммунодефицита, манифестированного частым развитием инфекционных заболеваний.
Рис. 4.16. Гигантский тромбоцит в мазке крови при аномалии Мей–Хегглина Приведено по: http://hematologia.narod.ru/Hematest1/test1_38.htm
145
|
Болезнь Вальденстрёма (сино- |
||
|
нимы – макроглобулинемия Валь- |
||
|
денстрёма, макроглобулинемия, мак- |
||
|
роглобулинемический |
ретикулёз, |
|
|
макроглобулинемическийретикуло- |
||
|
лимфоматоз). Болезнь органов кро- |
||
|
ветворения, характеризующаяся зло- |
||
|
качественной пролиферацией лим- |
||
|
фоидной ткани, |
сопровождающейся |
|
|
гиперпродукцией |
макроглобулинов |
|
|
(рис. 4.17). Проявляется прогрессиру- |
||
|
ющей слабостью, кровоточивостью |
||
|
слизистых оболочек, гепатоспленоме- |
||
|
галией, умеренным увеличением лим- |
||
|
фатических узлов, снижением зрения. |
||
|
Рентгенологически выявляются диф- |
||
|
фузный остеопороз, миелоподобные |
||
|
костные дефекты, бревиплатиспонди- |
||
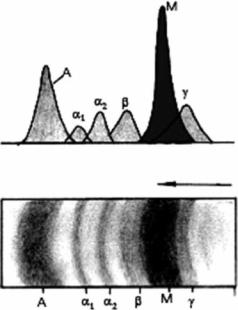
Рис. 4.17. Отчётливо выраженный пик ано- |
лия (снижение высоты и расширение |
||
мальных макроглобулинов с чрезвычайно вы- |
тела позвонка), плевральные шварты, |
||
соким молекулярным весом М-фракции, ко- |
сетчатое усиление лёгочного рисунка. |
||
торая обычно располагается в области между |
Описана в 1944 г. шведским вра- |
||
b- и g-глобулиновыми фракциями (ближе к зо- |
чом J. G. Waldenstrom. |
|
|
не g-фракции) (данные электрофореграммы |
Болезнь Шегрена (синоним – «су- |
||
белков крови при болезни Вальденстрёма) |
|||
Приведено по: Г. Е. Ройтберг, А. В. Струтын- |
хая болезнь»). Системное аутоиммун- |
||
ский (1999) |
ное заболевание, |
характеризующееся |
|
|
лимфоплазмоклеточной |
инфильтра- |
|
цией экзокринных желёз (главным образом слюнных и слёзных) с постепенным развитием их секреторной недостаточности, нередко – с вовлечением в патологический процесс опорно-двигательного аппарата, желудочно-кишеч- ного тракта, лёгких, почек, периферической и центральной нервной систем (рис. 4.18). Синдром Шегрена – тот же симптомокомплекс, но на фоне аутоиммунных заболеваний (ревматоидного артрита, системной склеродермии, системной красной волчанки, хронического активного гепатита, тиреоидита Хашимото). В развитии болезни не исключается роль цитомегаловирусов и вирусов Эпштейна–Барра. Генетическая детерминация заболевания подтверждается повышенной частотой HLA-DR3, -AB8, -DW2 и -DW 3. Самая распространённая теория патогенеза – аутоиммунная. Поражение слёзных желёз проявляется ксерофтальмией (снижением секреции слезы), жжением, чувством присутствия инородного тела в глазах, сухим нитчатым кератитом, конъюнктивитом, эпителиальной дистрофией роговицы. Развивается паренхиматозный паротит с ксеростомией (снижением продукции слюны) и выраженной сухостью во рту, вторичным возникновением хейлита, глоссита, при-
146
шеечного кариеса зубов, имеют место сухость кожи, половых органов, носа и глотки. Нередки бронхопневмонии, дискинезии кишечника, хронический холецистит, регионарная лимфаденопатия, синдром Рейно, нарушение функции почек (протеинурия, глюкозурия, аминоацидурия, гломерулонефрит) и рецидивирующие петехиальные высыпания на коже. Одним из самых серьёзных осложнений болезни и синдрома Шегрена является злокачественная лимфома.
Описана в 1933 г. шведским вра-
чом Н. С. Sjogren.
Концепция Бернета (синоним –
концепция иммунологического над-
зора). Специализированная система органов и клеток (иммунная система) отвечает за непрерывное выведение из организма генетически чужеродного материала. Она распознаёт «своё» и «чужое», даже если носитель этого «чужого» отличается всего лишь одним геном.
Выдвинута в 1971 г. австралийским учёным Ф. Бернетом.
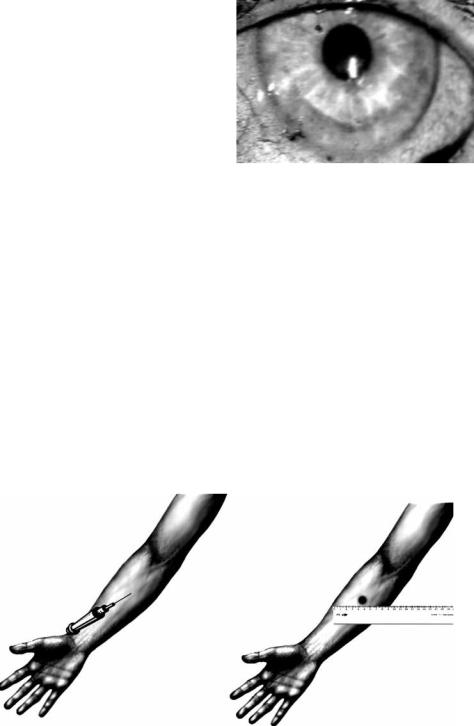
Проба Кавецкого (синоним – проба Кавецкого–Базарновой). Кожная проба для выявления неспецифической иммунологической резистентности организма человека. В область ладонной поверхности предплечья внутрикожно вводят 0,2 мл 0,25 %-ного раствора трипанового синего (рис. 4.19). Если
Рис. 4.19. Проба Кавецкого для оценки неспецифической иммунологической резистентности
147
через 24 ч после введения диаметр пятна менее 1 см, степень риска раневой инфекции у данного пациента достаточно высока.
Предложена в 1935 г. врачом Н. Е. Кавецким.
Проба Роттера. Кожная проба для выявления неспецифической иммунологической резистентности организма человека. В ладонную поверхность предплечья внутрикожно вводят 0,1 мл 0,1 %-ного раствора 2,6-дихлориндо- фенола (краски Тильманса) до формирования «лимонной корки». Время обесцвечивания краски более 15 мин указывает на снижение иммунологической резистентности организма и высокий риск развития инфекционных осложнений.
Реакция Пауля–Буннелля (синонимы – ХДПБ-реакция, реакция Хан-
гануциу–Дайхера–Пауля–Буннелля). Неспецифическая серологическая реакция гетерогемагглютинации эритроцитов барана, применяемая для ориентировочной диагностики инфекционного мононуклеоза.
Предложена в 1949–1952 гг. румынским иммунологом М. Hanganutziu, немецким врачом Н. Deicher, американскими врачами J. R. Paul и W. W. Bunnell.
Синдром Андерсона–Фридмана (синоним – LAD-синдром, или Leukocyte Adhesion Deficieincy I и II). Врождённое заболевание, характеризующееся первичным поражением системы фагоцитоза вследствие нарушения экспрессии молекул адгезии. У пациентов с LAD-I синдромом отсутствует или слабо выражена экспрессия CD18 на лейкоцитах, поэтому последние не могут прикрепляться к эндотелию и проходить в экстраваскулярное пространство. Исследование биопсийного материала, полученного из инфицированной ткани таких пациентов, выявляет полное отсутствие в ней нейтрофилов. У пациентов с LAD-II синдромом отсутствует экспрессия сиалил–ЛьюисХ-антигена на клетках эндотелия, являющегося лигандом для Р- и Е-селектинов фагоцитов, вследствие чего отсутствует роллинг фагоцитов и они не могут прикрепляться к эндотелию и проходить в экстраваскулярное пространство. Оба типа заболевания связаны с повышенным риском развития инфекций.
Внастоящее время сиалил-ЛьюисХ-антиген обозначен как антиген CD15s.
Кнему получены моноклональные антитела, которые могут быть использованы для диагностики этого заболевания.
Описан в 1992 г. врачами D. C. Anderson и М. Fridman.
Синдром Барандуна (синоним – идиопатическая гипогаммаглобукли-
немия). Заболевание, характеризующееся снижением концентрации гаммаглобулинов в сыворотке крови. Проявляется хроническими поражениями лёгких и кожи, гепато- и спленомегалией, иммуносупрессией. Предполагается, что в развитии синдрома ведущую роль играют генетические факторы.
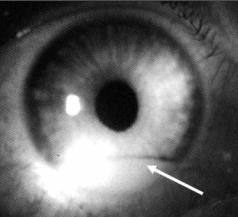
Синдром (болезнь) Бехчета. Заболевание, характеризующееся рецидивирующим афтозным (язвенным) поражением слизистых оболочек полости рта и наружных половых органов. Отмечаются поражения и других органов – глаз (увеит и его осложнения – гипопион, изменения хрусталика и стекловидного тела, слепота) (рис. 4.20), кожи (узловая эритема, узелки, папулы, пустулы,
148
иногда – язвы), суставов (артрит с поражением коленных, голеностопных, реже – лучезапястных и локтевых суставов, сакроилеит) и центральной нервной системы (атаксия, парезы, параличи, нарушения зрения и др.).
Относится к системным васкулитам иммунокомплексной природы. Считается достаточно редким заболеванием (хотя в Японии, Турции встречается часто). В развитии синдрома предполагается роль вируса простого герпеса I типа в сочетании с такими генетическими факторами, как связь с накоплением HLA B5 или HLA B 12,
а также DR7 BW52.
Описан турецким дерматологом Н. Behcet (1889–1948).
Болезнь (синдром) Брутона I
(синоним – первичная гипо- или агаммаглобулинемия взрослых). Наслед-
ственное заболевание семейного типа, сцепленное с Х-хромосомой (нередко наблюдается также рецессивный тип наследования). Отмечается склонность к повторным бактериальным инфекциям различной локализации (лёгкие, придаточные пазухи носа, кожа). Выявляется, как правило, уже в раннем дет - стве. К типичным признакам относятся диарея, а также увеличение периферических лимфатических узлов и селезёнки. Возможны системные ревматические проявления по типу диффузных болезней соединительной ткани. Суставной синдром характеризуется эпизодической мигрирующей полиартралгией либо острым, подострым, но чаще хроническим моноили асимметричным олигоартритом крупных суставов. В синовиальной жидкости признаки воспаления слабо выражены или отсутствуют вовсе. Даже при длительном течении артрит не приводит к рентгенологическим изменениям поражённых суставов. В анализах крови СОЭ в норме, острофазовые показатели не изменены, однако снижены уровни гаммаглобулинов (или они отсутствуют) и иммуноглобулинов (тотально или избирательно), отсутствуют изогемагглютинины. В костном мозге плазмоциты не выявляются или их содержание снижено; в биоптате лимфатического узла отмечается сужение кортикального слоя, первичные фолликулы в нём редкие и малоразвитые.
Описана в 1956 г. американским педиатром O. C. Bruton.
Синдром Вискотта–Олдрича (синоним – OMIM 301000). Первичное им-
мунодефицитное состояние Х-сцепленного типа (тяжёлый Х-сцепленный первичный иммунодефицит), встречающееся с частотой 0,4 случая на 100 000 новорождённых (рис. 4.21). Ген WAS, ответственный за развитие данного заболе-
149
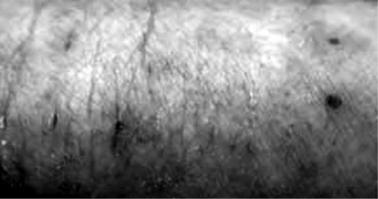
Рис. 4.21. Внешний вид кожи при атопическом дерматите на фоне синдрома Вискотта–Олдрича Приведено по: http://www.diagnos.ru/diseases/cutis/allerdermatit
вания, локализован на коротком плече Х-хромосомы в области Хр1122. ДНК-анализ Х-сцепленного синдрома позволяет осуществить прямую диагностику данного заболевания, способствуя установлению гетерозиготного носительства мутации у матери на раннем сроке беременности.
Синдром проявляется триадой симптомов, определяющихся у мальчиков с раннего возраста: 1) повышенной восприимчивостью к инфекционным заболеваниям (частые респираторно-вирусные и бронхолёгочные инфекции, инфекции ЛОР-органов, кожи, слизистых, мочевыводящих путей и ЖКТ); 2) геморрагическим синдромом, обусловленным тромбоцитопенией; 3) атопическим дерматитом.
Заболевание новорождённых обусловлено врождённым нарушением образования тромбоцитов и снижением содержания иммуноглобулинов крови. Локус WAS, расположенный на участке p11.22–p11.23 X-хромосомы, кодирует белок WASP с молекулярной массой 56 кДа, который выполняет адаптерные функции при внутриклеточной передаче сигнала во время агрегации тромбоцитов, индуцируемой тромбином, после их взаимодействия с волокнами коллагена. с-Концевая часть WASP взаимодействует с белковым комплексом Arp2/3, участвующим в перестройках цитоскелета, а также с GTPазой CDC42Hs-GTP, принадлежащей Rho-семейству, с последующим их транспортом в подосомы. Через белок CrcL WASP взаимодействует с тирозиновой протеинкиназой syk, являющейся основным участником передачи сигналов во время активации тромбоцитов коллагеном. Известно более 100 мутаций в гене WAS, большая часть которых изменяет аминокислоты N-концевой части белка WASP. Для данного синдрома характерны снижение уровней гемоглобина, эритроцитов, тромбоцитов, повышение уровня эозинофилов, изменение уровней сывороточных иммуноглобулинов (низкий IgM, нормальный IgG, высокий IgA, очень высокий IgE). Т-клеточные показатели вариабельны, и их интерпретация может быть затруднена.
150