

Ч 2
.pdfБолезнь (синдром) Стюарта–Прауэра. Врождённый геморрагический диатез, развивающийся вследствие дефицита фактора X свёртывания крови (тромбокиназа, КФ 3.4.21.6) и проявляющийся геморрагическим синдромом. Наследуется по аутосомно-рецессивному типу (ген F10 локализован на участке q34 хромосомы 13).
Названа в соответствии с фамилиями первых пациентов с данным заболе-
ванием – Stuart и Prower.
Болезнь Таруи (синонимы – гликогеноз VIII типа, миофосфофруктоки-
назная недостаточность). Гликогеноз, обусловленный врождённой недостаточностью или полным отсутствием активности фосфофруктокиназы в мышцах. Характеризуется мышечной слабостью, повышенной утомляемостью и отсутствием гиперлактацидемии после физической нагрузки.
Описана врачом S. Tarui.
Болезнь Томсона (синонимы – гликогеноз VII типа, гепатофосфоглюко-
мутазная недостаточность). Гликогеноз, обусловленный врождённой недостаточностью фосфоглюкомутазы в печени и/или мышцах. Проявляется метаболической миопатией, гепатомегалией.
Описана врачом W. Н. S. Thomson.
Болезнь Хага (синоним – гликогеноз IX типа ). Гликогенез, обусловленный врождённой недостаточностью киназы фосфорилазы В. Характеризуется гепатомегалией с отсутствием аппетита. Наследуется по рецессивному (сцепленному с полом) типу.
Описана врачом G. Hug.
Болезнь Ханса–Шюллера–Кристиана (синонимы – краниальный ксан-
томатоз, синдром Литтерера–Сайва, гистиоцитоз Х). В основе заболевания |
|
лежит нарушение обмена холестерина |
|
и инфильтрация липоидными и плаз- |
|
матическими клетками плоских ко- |
|
стей, твёрдой мозговой оболочки и ко- |
|
жи.Клиническиепризнаки:экзофтальм |
|
(рис. 7.12), половое недоразвитие, мел- |
|
кие желтоватые узелки в коже, пе- |
|
реломы длинных трубчатых костей, |
|
поражение лицевого нерва, снижение |
|
слуха, нистагм, мозжечковые рас- |
|
стройства. На рентгенограммах чере- |
|
па и других костей – очаги остеопоро- |
|
за неодинаковой интенсивности, с не- |
Рис. 7.12. Изменение периорбитальных тка- |
ровными контурами (напоминающие |
|
географическую карту). Заболевание |
ней при синдроме Ханса–Шюллера–Кристи- |
ана (данные МРТ черепа) |
|
начинается в возрасте до 10 лет. Чаще |
Приведено по: http://upload.wikimedia. org/wi- |
поражает мальчиков. Течение болезни |
kipedia/commons/thumb/8/8f/LCH1.jpg/180px- |
прогрессирующее. |
LCH1.jpg |
231
Описана австрийским врачом A. A. Schuller (1874–1958) и американским врачом H. A. Christian (1876–1951).
Болезнь (синдром) Хартнупа (синоним – Hartnup syndrome). Наслед-
ственная болезнь, обусловленная нарушением всасывания триптофана в кишечнике и его реабсорбции в почечных канальцах. Наследуется по рецессивному или аутосомно-рецессивному типу. Проявляется мозжечковой атаксией, пеллагроподобными изменениями кожи (тонкая, шелушащаяся кожа), аминоацидурией, повышенным содержанием в моче индикана и индольных соединений. Могут отмечаться нистагм, гиперрефлексия.
Описана врачом Е. Hartnup.
Болезнь Хатсусимы–Хакамори. Наследственная болезнь, обусловленная нарушением обмена полисахаридов с их накоплением в виде гранул в клетках паренхиматозных органов и в ретикулоцитах. Проявляется увеличением печени, селезёнки и лимфатических узлов, развитием цирроза печени.
Врождённые ошибки метаболизма Гаррода (синоним – congenital errors of metabolism). Случаи наследуемого нарушения способности осуществлять определённые ферментативные реакции.
Первые формы данных заболеваний были описаны А. Harrod в 1902–1908 гг. и носят его имя.
Клетки Ниманна–Пика. Крупные многоядерные ретикулоэндотелиальные клетки с пенистой протоплазмой, содержащей сфингомиелины. Обнаруживаются при болезни Ниманна–Пика.
Описаны немецким педиатром A. Niemann (1880–1921) и немецким патоло-
гоанатомом L. Pick (1868–1944).
Правило Уотсона. Правило, согласно которому наследуемые признаки обладают различной, характерной для каждого из них скоростью эволюции, в силу чего невозможно одновременное изменение большинства или всей совокупности признаков.
Описано американским учёным J. D. Watson.
Прогерия Гетчинсона (синонимы – синдром фатального старения, синдром раннего старения, синдром Гетчинсона–Гилфорда, прогерия Гар -
тингса). Редкая врождённая болезнь (частота возникновения 1 случай на 80 000 новорождённых), при которой происходит стремительное преждевременное старение организма. Дети, страдающие прогерией, могут выглядеть нормальными в течение 6–12 мес. после рождения, после чего у них развиваются симптомы, характерные для преклонного возраста, – морщинистая кожа, облысение, ломкость костей и атеросклероз. Как правило, такие дети гибнут в возрасте около 13 лет (рис. 7.13, 7.14). В развитии прогерии участвует ядерный протеин ламин А. У детей, страдающих прогерией, в результате мутации отсутствует участок молекулы ламина А, включающий критическую точку, по которой происходит отрезание модифицированного участка (хотя теоретически существует множество возможных вариантов формирования патологического ламина А).
232

Рис. 7.13. Внешний вид пациентов с прогерией Гетчинсона–Гилфорда Приведено по: http://www.scientific.ru/journal/news/1005/n261005.html
Рис. 7.14. Внешний вид новорождённого и изменения кожи при синдроме Гетчинсона–Гилфорда
Приведено по: http://www.scientific.ru/ journal/news/1005/n261005.html
Описана английскими врачами J. Hutchinson (1828–1913) и H. Gilford (1861– 1941).
Синдром Абдергальдена–Кауфманна–Линьяка (синонимы – цистиноз, синдром Абдергальдена–Фанкони, диабет гликофосфаминный, синдром Линьяка, синдром Линьяка–Фанкони). Наследственная болезнь, обуслов-
ленная нарушением обмена цистина с отложением его кристаллов в ретикулярных клетках костного мозга, печени, селезёнки и лимфатической системы, а также в клетках роговицы и конъюнктивы. Наследуется по рецессивному типу. Характеризуется задержкой роста, развитием рахитоподобных изменений в костях, нарушением зрения, аминоацидурией, глюкозурией, гипофосфатемией и анемией.
233
Описан швейцарским биохимиком и физиологом Е. Abderhalden (1877– 1950), немецким патологоанатомом Е. Kaufmann (1860–1931) и голландским патологоанатомом G. О. Е. Lignac (1891–1954).
Синдром (болезнь) Александера(синонимы – SPCA deficiency, Alexander syndrome, гипоконвертинемия, дефицит фактора VII). Наследственная аномалия свёртывания крови, обусловленная дефицитом фактора VII свертывания (акселератор конверсии сывороточного протромбина). Болезнь поражает лиц обоего пола. Передается по аутосомно-рецессивному типу (ген F7 локализован на участке q34 хромосомы 13). Проявляется после рождения в виде кровотечения из пуповины, пищевого канала и внутричерепных кровоизлияний. В более позднем возрасте проявляется в виде носовых, маточных, десневых, посттравматических кровотечений, а также внутрисуставных кровоизлияний (напоминает гемофилию).
Описан врачом В. Alexander.
Синдром Альстрема–Ольсена. Врождённое заболевание с аутосомно-ре- цессивным типом наследования. К основным его проявлениям наряду с сахарным диабетом и ожирением относятся атипичная пигментная дегенерация сетчатки, нейросенсорная глухота и нефропатия.
На первом году жизни появляются нистагм и светобоязнь, а также ретинит. Постепенно прогрессирует снижение центрального и периферического зрения, может развиться слепота. С раннего детства формируется ожирение. В юношеском возрасте становится заметным снижение слуха (вплоть до полной глухоты). После пубертатного периода появляются признаки инсулиннезависимого сахарного диабета, который часто осложняется нефропатией, приводящей к хронической почечной недостаточности. Половое развитие чаще нормальное, иногда наблюдается мужской гипогонадизм. При биопсии яичек обнаруживают аплазию герминативных клеток и склероз семенных канальцев. Интеллект обычно сохранен. У мальчиков заболевание отмечается вдвое чаще. Прогноз заболевания неблагоприятный.
Описан в 1959 г. шведскими офтальмологами С. Н. Alstrom и О. Olsen.
Синдром (болезнь) Андерсена (синонимы – гликогеноз IV типа, амило-
пектиноз). Редкое врождённое (семейное) заболевание, проявляющееся отложением в печени аномального полисахарида (сходного по своим свойствам с амилопектином). Причина – врождённое отсутствие ветвящего фермента 1,4-альфа-глюкозил-трансферазы. Многие аспекты возникновения болезни и механизмы нарушения синтеза ветявящего фермента изучены недостаточно. Характеризуется прогрессирующей гепатоспленомегалией, возникновением цирроза печени, атрофией поджелудочной железы. В большинстве наблюдений заболевание протекает без гипогликемии (иногда с поликистозной фиброзной трансформацией поджелудочной железы). Сразу после рождения выявляются увеличение живота, усиленное выделение жирного кала.
В диагностике важно обнаружение повышенного уровня ветвящего фермента в коже (активность фермента отсутствует в печени, почках, селезёнке,
234

Рис. 7.16. Внешний вид ребёнка с синдромом Берьесона (ожирение, гипотиреоз, олигофрения, задержка роста)
Приведено по: Н. Б. Ситковский с соавт. (1981)
гофренией (рис. 7.16). Наследуется по рецессивному, сцепленному с полом (передача повреждённой Х-хромосомы), типу. С рождения отмечается мышечная гипотония, выраженная задержка развития. Дети начинают ходить в 4–5 лет. Характерны отставание в росте, умеренное ожирение (с возрастом уменьшается), тяжёлая умственная отсталость, выраженная задержка речевого развития. Черепно-лицевые аномалии включают микроцефалию, грубые черты лица с выступающими надбровными дугами и запавшими глазами. Отмечается отёчность тканей лица, приводящая к сужению пространства между веками (иногда веки приходится поднимать с помощью пальцев). Наблюдаются нистагм, снижение зрения вследствие поражения сетчатки и зрительных нервов. Отмечаются микропения, задержка появления вторичных половых признаков, гипогонадотропный гипогонадизм, а также идиотия, эпилепсия, гипотиреоз (иногда с явлениями микседемы), увеличение в размерах (без деформации) ушных раковин. Рентгенологически выявляются утолщение костей черепа, умеренный сколиоз или кифоз, аномалии позвонков, расширение метафизов длинных трубчатых костей и костей кисти, гипоплазия дистальных и средних фаланг.
Описан в 1961 г. врачами М. Borjeson, J. Forssman и О. Lehmann.
Синдром Бессена–Корнцвейга (синонимы – абеталипопротеинемия,
акантоцитоз). Наследственная болезнь, характеризующаяся нарушением всасывания и транспорта жиров, недостаточностью или отсутствием бета-липо- протеинов, недостаточностью высших полиненасыщенных жирных кислот и низким содержанием холестерина и фосфатидов в крови. Наследуется по ау- тосомно-рецессивному типу. Проявляется акантозом, пигментной дегенерацией сетчатки (рис. 7.17), атаксией, наличием в крови акантоцитов.
236
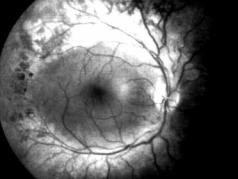
Описан в 1968 г. американскими врачамиF.A.BassenиA.L.Kornzweig.
Синдром Бломстренда (синоним –
синдром летальной хондродисплазии Бломстренда). Врождённое забо-
левание с аутосомно-рецессивным типом наследования и ранним летальным исходом. Новорождённые с данным синдромом, как правило, имеют здоровых родителей, состоящих в близкородственном браке. Идентифицированы мутации гена РТН/PTHrP рецептора, которые и обусловливают нарушения функции рецептора с формированием фенотипа болезни (ген
PTHR/PTHrPR локализован на участке 3р21.1–р22). Болезнь характеризуется повышенным созреванием костей и ускоренной дифференцировкой хондроцитов. Быстро развивается избыточная минеральная плотность скелета, отмечается ускоренная оссификация костей с формированием грубых нарушений строения длинных трубчатых костей и очень низким ростом. Новорождённые с синдромом летальной хондродисплазии Бломстренда имеют и другие аномалии – аплазию соска и молочной железы. Регистрируется вторичная гиперплазия паращитовидных желёз из-за резистентности к паратгормону и ПТГподобному пептиду (РТНrР).
Впервые описан шведским учёным К. W. Blomstrand (1826–1897).
Синдром Бродбергер–Цеттерстрёма. Врождённое заболевание с ауто-
сомно-рецессивным типом наследования. Отмечается дефицит гликогеназы печени, выявляется гипогликемия. Уровень адреналина под влиянием гипогликемии, обусловленной инсулином, не меняется.
Описан английским врачом W. Brodberger (1868–1951).
Синдром (болезнь) Брутона II (синоним – сцепленная с полом агамма -
глобулинемия). Врождённое заболевание, связанное с Y-хромосомой (поражает только мальчиков). Проявляется рецидивирующими и тяжело протекающими инфекциями и артропатиями. Пейеровы бляшки на слизистой оболочке тонкой кишки, миндадины и аппендикс отсутствуют.
Описан в 1957 г. американским педиатром O. C. Bruton.
Синдром Ван-Вика–Грамбаха (синоним – синдром гиперпролактинемического гипогонадизма при первичном гипотиреозе). Форма первичного гипотиреоза. Заболевание развивается в допубертатном периоде. Характеризуется преждевременным половым созреванием, галактореей, преждевременным менархе с метроррагиями при отсутствии полового оволосения. Нарушение менструального цикла вплоть до аменореи, реже – полименореи. Аналогичный мужской вариант синдрома характеризуется ускоренным развитием
237
наружных половых органов при отсутствии или скудости полового оволосения. Нередким является сочетание с болезнью Дауна. Частыми находками у детей с синдромом Ван-Вика–Грамбаха являются гиперпролактинемия и аденома гипофиза. Характерна макромастия. Галакторея обычно более выражена у молодых женщин. Её степень варьируется от обильного и спонтанного выделения молока до появления единичных капель при сильном надавливании на молочные железы. В последнем случае сами пациентки, как правило, галактореи не отмечают. В молочных железах (которые внешне увеличены в размерах) часто наблюдаются не соответствующие возрасту регрессивные изменения, фиброзно-кистозная мастопатия. Характерны снижение либидо, аноргазмия, атрофия матки и слизистой оболочки влагалища. Нередко выявляются умеренное ожирение, гипертрихоз, себорея волосистой части головы, акне и признаки дисфункции гипоталамуса («грязные» локти и шея вследствие гиперкератоза и гиперпигментации, перламутровые стрии на бёдрах, груди и животе).
Описан в 1959 г. американским эндокринологом J. J. Van Wyk и американ - ским педиатром M. M. Grumbach.
Синдром Вермера (синонимы – множественная эндокринная неоплазия I типа или МЭН I типа, випома, РРома, WDHH синдром, псевдохолера,
панкреатическая холера). Обусловленное генетическими дефектами заболевание, наследуемое по аутосомно-доминантному типу. Для него характерны:
а) гиперпаратиреоз (встречается более чем у 90 % пациентов и обусловлен гиперплазией нескольких паращитовидных желёз; поражения почек и костей наблюдаются не всегда);
б) опухоли аденогипофиза (отмечаются более чем у 50% пациентов; обычно эти опухоли не секретируют гормоны; активно растущие злокачественные опухоли сдавливают и разрушают гипофиз и приводят к гипопитуитаризму; реже встречаются аденомы, секретирующие пролактин, СТГ или АКТГ, вызывающие галакторею и аменорею, акромегалию или гипофизарный синдром Кушинга);
в) опухоли из островковых клеток (имеются более чем у 50 % пациентов; обычно они множественные и происходят как из бета-клеток, так и из других островковых клеток; наиболее распространены инсулиномы, глюкагономы, гастриномы и випомы);
г) опухоли надпочечников (у 40 % пациентов имеется гиперплазия или аденома надпочечников; гиперсекреции глюкокортикоидов обычно нет, поэтому гиперплазию и аденому обнаруживают случайно либо при детальном обследовании);
д) заболевания щитовидной железы (имеются примерно у 20 % пациентов; обычно это аденомы или злокачественные опухоли, реже встречаются тиреотоксикоз, коллоидный зоб и хронический лимфоцитарный тиреоидит).
Достаточно редко при данном синдроме выявляются рак лёгкого, липомы, полипоз желудка, опухоли яичек и шванномы. Установлено, что причиной
238
МЭН I типа служат мутации гена супрессии опухолей (11q13). Этот ген кодирует белок мэнин, регулирующий пролиферацию клеток – производных нервного гребня. Недостаточность мэнина приводит к гиперплазии, а его отсутствие – к опухолевой трансформации этих клеток. Ещё одна предполагаемая причина МЭН I типа – повышенный уровень фактора роста фибробластов в сыворотке (известно, что фактор роста фибробластов стимулирует пролиферацию клеток паращитовидных желёз).
Заболевание чаще всего проявляется между 20 и 40 годами, но может возникнуть и до 10 лет. Первым проявлением МЭН I типа обычно бывает гиперпаратиреоз. При наличии инсулиномы отмечается гипогликемия. Симптомы глюкагономы – гипергликемия и некролитическая мигрирующая эритема. Гастриномы проявляются синдромом Золлингера–Эллисона (пациенты с этим синдромом страдают язвенной болезнью и другими нарушениями ЖКТ). Проявление випомы – панкреатическая холера (водянистый стул, гипокалиемия, ахлоргидрия, метаболический ацидоз, эксикоз, кахексия, гиповолемический шок, тахикардия, гипотония, мышечная слабость, атония ЖКТ). Это связано с тем, что вазинтестинальный полипептид (ВИП) активирует интестинальную и панкреатическую аденилатциклазу – усиливает секрецию поджелудочной железы и тонкой кишки (действие, схожее с действием холерного токсина, вызывающего профузную диарею). Как правило, в каждой эндокринной железе изначально имеется несколько очагов гиперплазии или опухолевого роста (поскольку любая эндокринная клетка потенциально способна к трансформации). Вначале возникает гиперплазия, затем доброкачественная и, наконец, злокачественная опухоль (поэтому заболевание развивается постепенно и может не проявляться до 35–40 лет). Клиническая картина и лабораторные показатели зависят от того, на какой стадии развития опухоли выявлено заболевание.
Впервые описан в 1954 г. врачом Р. Wermer.
Синдром Вернера II (синонимы – прогерия взрослых, ангидратиче-
ский синдром). Наследственное заболевание человека, характеризующееся врождёнными эндокринными расстройствами. По патогенезу оно близко к синдрому Ротмунда (в частности, приводит к преждевременному старению) (рис. 7.18). Наследуется по аутосомно-рецессивному типу. Проявляется обычно в возрасте 20–30 лет. Чаще страдают лица мужского пола. Характерными признаками являются задержка роста, раннее появление седых волос, облысение, выпадение зубов, склеродермия (особенно выраженная на конечностях), атрофия подкожно-жирового слоя и мышечной ткани, сахарный диабет и гипогонадизм. Кожа становится бледной, истончённой, несколько уплотнённой, лицо заостряется, становится сморщенным, с клювовидным носом. Развивается двусторонняя катаракта, макулярная дегенерация, пигментный ретинит, хориоретинит. Рентгенологически выявляются признаки остеоартроза, деформации костей стоп, остеоартриты мелких суставов рук и ног. Наблюдаются признаки генерализованного атеросклероза, стенокардия и инфар-
239

|
кты миокарда. Нередко возникают |
|
злокачественные опухоли. Прогноз |
|
заболевания неблагоприятный. |
|
Описан в 1906 г. врачом О. Werner. |
|
Синдром Видемана–Беквита (си- |
|
нонимы – синдром омфалоцеле, син- |
|
дром макроглоссии, синдром гиган- |
|
тизма, EMG-синдром). Врождённое |
|
заболевание с аутосомно-доминант- |
|
ным типом наследования. В некото- |
|
рых случаях выявляют структурные |
|
перестройки 11-й хромосомы. Назва- |
|
ние «EMG-синдром» происходит из |
|
трёх наиболее манифестных его при- |
|
знаков – экзомфалоса (E), макроглос- |
|
сии (M) и гигантизма (G). У ново- |
|
рождённых отмечается гипогликемия |
Рис. 7.18. Синдром Вернера II у пациента |
в сочетании с соматическими измене- |
38 лет |
ниями. |
Приведено по: http://zhurnal.lib. ru/img/m/ |
Основными клиническим призна- |
melxnik_anatolij_antonowich/corp-idoc/corp- |
ками являются макроглоссия, пупо- |
1-i.jpg |
винная грыжа (рис. 7.19) и другие пу- |
|
|
|
почные аномалии, а также гипоглике- |
мия. Макроглоссия выявляется в любом возрасте ребёнка, часто – с рождения. Язык может не помещаться во рту, поэтому рот ребёнка открыт, а лицо напоминает лицо пациента с гипотиреозом. Увеличенный язык затрудняет сосание и даже дыхание новорождённого. У детей более старшего возраста отмечают дизартрические расстройства. Гипогликемия у новорождённого манифестирует уже на первые-третьи сутки. Развивающиеся за счёт этого коматозные состояния могут повлечь за собой смерть ребёнка на первом году жизни или тяжёлое органическое поражение мозга, сопровождающееся умственной отсталостью. Развитие гипогликемических состояний связано с гиперплазией островковых клеток поджелудочной железы, приводящей к гиперинсулинемии. С начинающейся ещё внутриутробно гиперпродукцией инсулина, обладающего анаболическим действием, связывают как макроглоссию, макросомию, висцеромегалию, так и предрасположенность к развитию опухолей паренхиматозных органов. Явления гипергликемии самопроизвольно убывают в течение первых месяцев жизни пациента. Склонность к увеличению массы тела (обычно она превышает 4000 г) отмечается уже при рождении, при этом длина тела составляет 52 см. Внутриутробная висцеромегалия, по-видимому, является причиной образования различных грыж, в том числе характерного для синдрома омфалоцеле (пуповинной грыжи). Пуповинная грыжа диагностируется у новорождённого и может быть различной по величине, достигая
240