

Ч 2
.pdfство жизни – смерть, как правило, наступает от пневмонии. Примерно 75 % пациентов умирает до 20 лет, 5 % доживает до 50-летнего возраста и более. Для диагностики заболевания важно определение в крови активности креатинфосфокиназы (у пациентов она повышена). В биоптате мышц выявляют патологические волокна, окружённые жировой и фиброзной тканью.
Описана французским невропатологом
G. В. А. Duchenne (1806–1875).
Болезнь Канавана (синонимы – спонгиозная дегенерация белого вещества мозга, губчатая дегенерация мозга, болезнь Кана-
вана–Ван-Богарта–Бертрана, болезнь Ка-
навана). Наследственная болезнь, обусловленная прогрессирующей демиелинизацией белоговеществаголовногомозгасобразованием полостей (рис. 7.52). Форма лейкодистрофии
характеризуется недостатком фермента аспартоацилазы и повышенным со- держаниемвмоче,плазмеикостноммозгеметаболитаN-ацетиласпарагиновой кислоты. Передаётся по аутосомно-рецессивному типу. Проявляется адинамией, судорогами, гиперкинезами и симптомами поражения черепных нервов. Летальность наступает в раннем возрасте.
Описана американским врачом М. М. Canavan (1879–1953).
Болезнь Ли II (синоним – подострая некротизирующая энцефаломие-
лопатия). Наследственная болезнь из группы демиелинизирующих заболеваний, характеризующаяся прогрессирующей дистрофией белого вещества головного и спинного мозга и проявляющаяся нарастающими пирамидными и экстрапирамидными расстройствами, нарушением функций черепных нервов, поражением миокарда.
Описана в 1949 г. английским невропатологом D. Leigh.
Болезнь Паркинсона (синоним – дрожательный паралич). Наследствен-
ное заболевание человека (рис. 7.53), характеризующееся тремором (дрожанием) и другими проявлениями поражений головного мозга. Передаётся предположительно по аутосомно-доминантному типу (с низкой пенетрантностью).
Описана английским врачом J. Parkinson (1755–1824).
Болезнь Ферраро–Ван-Богарта–Ниссена. Семейная болезнь централь-
ной нервной системы, характеризующаяся атрофией коры полушарий большого мозга и дегенерацией базальных ядер. Проявляется в возрасте 40–45 лет развитием деменции и акинетико-ригидного синдрома.
Впервые описана в 1937 г. бельгийскими врачами A. Ferraro и L. van Bogaert.
281

Рис. 7.53. МРТ головного мозга при различных стадиях болезни Паркинсона Приведено по: http://www.nature.com/clpt/journal/v81/n1/images/6100013f5.jpg
Киста Верги (синоним – киста прозрачной перегородки). Аномалия раз-
вития – скопление жидкости между листками прозрачной перегородки головного мозга.
Впервые описана итальянским невропатологом и психиатром A. Verga (1811–1895).
Наследственная атаксия Фридрейха (синоним – семейная атаксия Фридрейха). Наследственная патология, характеризующаяся прогрессирующими нарушениями координации движений (атаксией), врождёнными пороками сердца, деформациями скелета (позвоночника, грудной клетки, стоп и др.), дистрофическими изменениями миокарда. Наследуется по аутосомнодоминантному или аутосомно-рецессивному типу. Локус FRDA расположен на участке q13–q21 хромосомы 9.
Описан немецким врачом N. Friedreich (1825–1882).
Нейропатия Лебера (синонимы – синдром Лебера, «врождённая ошибка метаболизма» с нарушением детоксикации цианида, атрофия зритель-
ного нерва Лебера). Врождённое заболевание, наследуемое по материнской линии (по рецессивному, сцепленному с полом, типу). Синдром обусловлен
282

Рис. 7.54. Изменения при нейропатии Лебера: а – нарушение полей зрения ( 1 – слепое пятно; 3 – центральная скотома);б – атрофия зрительного нерва (вид глазного дна при офтальмоскопии)
Приведено по: http://medarticle.moslek.ru/images/18040.jpg, http:// medarticle. moslek.ru/ images/13909.jpg
мутациями в гене IV субъединицы НАДФ-коэнзим-Q-редуктазы, входящем в состав митохондриального генома. Характеризуется прогрессирующими нарушениями зрения в юношеском возрасте (чаще поражает лиц мужского пола) и проявляется острой или подострой двусторонней потерей зрения с образованием центральной скотомы (рис. 7.54).
Описана немецким офтальмологом Th. Leber (1840–1917).
Симптом (феномен) Штрассбургера. Диафаноскопия черепа положи-
тельна в крайних случаях гидроцефалии.
Описан врачом J. H. Strassburger.
Синдром Алана. Врождённая патология неизвестной этиологии, характеризующаяся задержкой умственного развития (разновидность дисметаболической олигофрении) в раннем детском возрасте. Сочетается с эпилептическими припадками и атаксией.
Синдром (болезнь) Альцгеймера (синоним – пресинильное слабоумие).
Наследственное заболевание человека, характеризующееся прогрессивным слабоумием – нарушениям памяти, утратой навыков (например, навыков письма), психотическими расстройствами, гиперкинезами и т. п., что связано с диффузной атрофией мозга (рис. 7.55). Морфологически от старческой церебральной атрофии отличается большим числом сенильных бляшек и нейрофибриллярных клубочков (сплетений), являющихся патологоанатомическими маркёрами деменции Альцгеймера. Непрерывно прогресссирующее течение также отличает её от ступенчатых ухудшений на фоне мультиинфарктной деменции. Проявляется после 55 лет (но может наблюдаться и в более молодом возрасте), 70 % больных – женщины.
283

|
Для первой стадии характерны |
||
|
потеря |
памяти и пространственная |
|
|
дезориентация, во второй стадии про- |
||
|
исходит распад личности (с нараста- |
||
|
нием агрессивности) и появляются |
||
|
очаговые изменения со стороны те- |
||
|
менной доли мозга (дисфазия, апрак- |
||
|
сия, агнозия и акалькулия), в третьей |
||
|
стадии пациент становится апатич- |
||
|
ным (или чрезмерно активным – ака- |
||
|
тизия), у него снижается масса тела, |
||
|
он прикован к постели, возникают та- |
||
|
зовые |
расстройства (недержаниие |
|
|
мочи и кала), в последней стадии ча- |
||
Рис. 7.55. Изменения со стороны головного моз- |
сто отмечаются судорожные припад- |
||
ки и спастичность мышц. Наследует- |
|||
га при болезни Альцгеймера (б) по сравнению |
|||
с головным мозгом здорового человека (а) |
ся по аутосомно-доминантному типу |
||
Приведено по: http://www.alzheimer.ca/english/ |
(локус AD1 расположен на участке |
||
alzheimer_brain_mini_site/images/09a.jpg |
q21.1 хромосомы 21). Средняя продол- |
||
жительность жизни с момента начала болезни около 7 лет.
Описан немецким врачом A. Alzheimer (1864–1915).
Синдром Арнольда–Киари (синонимы – синдром Арнольда–Киари–
Соловцова, Arnold–Chiari malformation). Краниовертебральная мальфор-
мация – сочетание атаксии, нистагма, параличей черепных нервов и эпилептиформных судорог, обусловленное врождённым смещением головного мозга в каудальном направлении с ущемлением его в увеличенном (расширенном) затылочном отверстии (рис. 7.56). Обычно сопровождается дефектами развития нервной трубки с нарушениями ликвородинамики и гидроцефалией. Аномалия развития центральной системы относится к так называемым мультифакторным порокам, т. е. причиной его является наследственная предрасположенность у супругов (мутации генов, участвующих в обмене фолиевой кислоты) в сочетании с неблагоприятным внешним воздействием в 2–3 недели беременности. Риск повторного появления на свет ребёнка с таким же дефектом примерно 3–5 %.
Характерная для заболевания инфантильная конституция пациентов связана со спинальными дизрафическими аномалиями. Проявляется в виде гидроцефалии, поражения мозжечка, компрессионных поражений ствола мозга и спинного мозга, нередко в сочетании с аномалией шейных позвонков, син-
дромом Клиппель–Фейля, синдромом Киммерли. Отмечаются головокру-
жение и/или шаткость (которые могут усиливаться при повороте головы); шум (звон, гул, свист, шипение и т. п.) в одном или обоих ушах (может усиливаться при повороте головы); головная боль, связанная с повышением внутричереп-
284

ного давления (сильнее утром) или с повышением тонуса мышц шеи (болевые точки под затылком); нистагм (непроизвольное подёргивание глазных яблок). В особо тяжёлых случаях возможны преходящая слепота, двоение или другие зрительные расстройства (как правило, при повороте головы); тремор рук, ног, расстройство координации движений; снижение чувствительности части лица, части туловища, одной или нескольких конечностей; слабость мускулатуры части лица, части туловища, одной или нескольких конечностей; непроизвольное или затруднённое мочеиспускание; потеря сознания (может провоцироваться поворотом головы). Рентгенологически отмечается сужение большого затылочного отверстия.
Описан немецким патологом J. Arnold в 1894 г. и австрийским мор-
фологом Н. Chiari в 1895 г.
Синдром Аспергера. Врождённая аутистичная психопатия. Проявляется аутизмом, при котором ребёнок с 2 лет
навязчиво повторяет одни и те же слова или настойчиво занимается одним
итем же «делом», тяготеет к стереотипическому поведению. Изменение обстановки становится для него тяжелейшим стрессом. При этом у больного ребёнка отсутствуют самые главные черты аутизма – аутистическая уединённость (ребёнок не в состоянии отвечать на те или иные чувства родителей)
илингвистические трудности (бедность речи, неправильное употребление местоимений, неадекватное повторение слов). Прогноз более благоприятный, чем при классическом аутизме.
Термин ввёл в обиход в 1981 г. L. Wing в честь австрийского психиатра
ипедиатра H. Asperger, который сам для трактовки этого заболевания использовал термин «аутистическая психопатия».
Синдром Беккера (синонимы – Becker syndrome, X-хромосомная псевдогипертрофическая доброкачественная миопатия). Рецессивное (сцеплен-
ное с полом) наследственное нервно-мышечное заболевание. От мышечной дистрофии Дюшенна отличается менее выраженным прогрессивным течением, более поздним началом и значительным увеличением икроножных мышц в подростковом возрасте.
Описан врачом Р. Е. Becker.
285

Синдром (болезнь) Дежерина–Сотта (синонимы – гипертрофический неврит Дежерина–Сотта, гипертрофическая семейная невропатия, интерстициальный гипертрофический прогрессирующий полиневрит). Наслед-
ственная болезнь, характеризующаяся гипертрофией шванновской оболочки нервных волокон со сдавлением и дегенерацией аксонов (рис. 7.57). Проявляется медленно нарастающими периферическими парезами с мышечными атрофиями и нарушениями чувствительности по полиневритическому типу. Наследуется по аутосомно-рецессивному типу.
Описан французскими невропатологами J. J. Dejerine (1849–1917) и J. Sottas (1866–1924).
Синдром Джонстона (синонимы – синдром зеркального движения рук,
Johnstone syndrome). Врождённая аномалия развития центральной нервной системы – неполный перекрёст волокон пирамидного пути на границе спинного и продолговатого мозга. Проявляется зеркальными движениями рук (при попытке пациента сделать движение одной рукой вторая в точности повторяет это движение).
Описан врачом J. Johnstone.
Синдром Киммерли. Краниовертебральная мальформация – дополнительная костная дужка первого шейного позвонка (рис. 7.58), которая может сдавливать позвоночные артерии, питающие головной мозг кровью. Нередко сочетается с синдромом Арнольда–Киари.
Для нормальной работы головного мозга требуется большое количество питательных веществ, поэтому головной мозг человека обеспечен мощной системой артерий, подающих кровь для его питания. Задние отделы мозга пита-
Рис. 7.57. Гипертрофия шванновской оболочки с дегенерацией нервного ствола при синдроме Дежерина–Сотта. ×5000
Приведено по: http://www.anestesiarianimazione.com/Immagini/a06f02.gif
286

Рис. 7.58. Рентгенограмма пациента и механизм сдавления позвоночной артерии в костной дужке-полукольце (а – нормальный позвонок; б – ущемление артерии в аномальном пулокольце) при синдроме Киммерли
Приведено по: http://images.nature.web. ru/nature/2001/10/22/0001172560/pic2. jpg
ются главным образом из системы позвоночных артерий, проходящих в полость черепа прямо через шейные позвонки. Позвоночные артерии (правая
илевая) огибают первый шейный позвонок, затем проходят в полость черепа, где и включаются в общую систему мозгового кровообращения. При аномалии Киммерли над дугой первого шейного позвонка имеются аномальные дополнительные костные дужки-полукольца, которые могут сдавливать позвоночные артерии. Сдавление артерии происходит между аномальной дужкой
имышцами шеи при некоторых положениях головы или избыточном напряжении шейных мышц. Сдавление позвоночных артерий приводит к дефициту кровоснабжения головного мозга. Болезнь проявляется головокружением и/или шаткостью (может усиливаться при повороте головы); шумом (звоном, гулом, свистом, шипением т. п.) в одном или обоих ушах (может усиливаться при повороте головы); потемнением в глазах, «мушками» или «звёздочками» перед глазами при напряжении или некомфортном положении мышц шеи, повороте головы; утратой сознания (полной или частичной) при напряжении или некомфортном положении мышц шеи, повороте головы; внезапной слабостью мышц и падением при сохранённом сознании. В особо тяжёлых случаях отмечаются головная боль, нистагм (непроизвольное подёргивание глазных яблок), тремор рук и ног, расстройства координации движений, снижение чувствительности участков лица, части туловища, одной или нескольких конечностей, слабость мускулатуры лица, части туловища, одной или нескольких конечностей.
Синдром Клиппеля–Фейля. Краниовертебральная мальформация – сра-
щение 2–3 шейных позвонков между собой (рис. 7.59). Часто приводит к сдавлению позвоночных артерий, перегрузке и повреждению выше- и нижележащих межпозвонковых дисков. Встречается с частотой 1 случай на 42 000 ново-
287
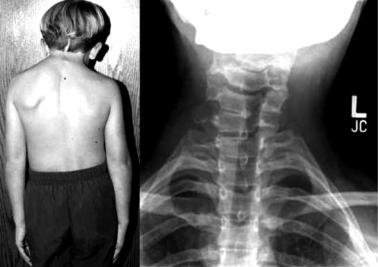
Рис. 7.59. Внешний вид и рентгенограмма шеи пациента с синдромом Клиппеля–Фейля
Приведено по: http://www.werathah.com/klippel/imagies/head3.jpg, http://www.mypacs.net/repos/ mpv3_repo/viz/full/55045/2752270.jpg
рождённых. В 10 % случаев синдром сочетается с другими пороками развития опорно-двигательного аппарата, чаще – с синдромом Арнольда–Киари. Клинически нередко проявляется как синдром Киммерли или синдром Арноль- да–Киари.
Рентгенологически выделяют два типа заболевания: 1) атлант и эпистрофей соединены с остальными позвонками (числом не более четырёх), дужки не заращены, сзади первые рёбра присоединены выше, чем спереди; 2) синостоз атланта с затылочной костью, низлежащие позвонки соединены между собой, могут быть добавочные полупозвонки и шейные рёбра.
Описан в 1929 г. французскими невропатологами М. Klippel (1858–1942)
и A. Feil.
Синдром Кобба. Врождённый ангиоматоз спинного мозга и кожи. Описан врачом S. Cobb.
Синдром Лендузи–Дежерина (синоним – мышечная дистрофия Ленду-
зи–Дежерина). Заболевание, наследуемое по аутосомно-доминантному типу. Проявляется слабостью мышц плечевого пояса (пациенту трудно причесаться гребнем). Появляется обычно в 12–14 лет. Пациент также испытывает трудности при закрывании глаз, сосании, при попытке что-либо надуть или засвистеть, лопатки опущены, губы «надуты», а выражение лица как бы «отутюженное» (миопатическое лицо). Количество взрослого миоглобина уменьшено, а «фетального» миоглобина и ядер в сарколемме мышечных клеток увеличено.
Впервые описан французским невропатологом J. J. Dejerine (1849–1917).
288

Синдром Ленобля–Обино (синоним – нистагм-миоклония). Наследствен-
ная болезнь, характеризующаяся врождённым нистагмом, спонтанными фасцикулярными подёргиваниями мышц, тремором и вазомоторными расстройствами.
Впервые описан французским врачом Е. A. Lenoble (1864–1928).
Синдром Маринеску–Шегрена (синонимы – синдром Маринеску–Дра- ганеску–Василиу, синдром Маринеску–Шегрена–Гарленда). Наследствен-
ная болезнь, характеризующаяся сочетанием мозжечковой атаксии, катаракты и олигофрении. Наследуется по аутосомно-рецессивному типу.
Впервые описан румынским невропатологом G. Marinescu (1863–1938). Синдром Мёбиуса. Наследственное заболевание человека, характеризующееся параличом черепных нервов (VI и VII пары), приводящим к маскообразному виду лица, косоглазию и гипомимии. Часто наблюдаются деформации конечностей. Наследуется по аутосомно-доминантному типу (локус
синдрома MBS расположен на участке q12.2 хромосомы 13). Описан немецким невропатологом P. J. Moebius (1853–1907).
Синдром (болезнь) Пика II (синоним – деменция Пика). Врождённое за-
болевание, проявляющееся позже, чем болезнь Альцгеймера. Поведение пациентов полностью укладывается в понятие «лобной психики» (ослабление внутреннего самоконтроля – грубая бестакность, тяга к воровству и необычным сексуальным похождениям). Приличное поведение в обществе заменяется глупой самодовольной эйфорией и жаргонной дисфазией. Все эти изменения наблюдаются до того, как происходит снижение интеллектуального и неврологического статуса (рис. 7.60).
Описан немецким невропатологом L. Pick (1868–1944).
Синдром (болезнь) Пьера Мари (синонимы – наследственная мозжечковая атаксия Пьера Мари, наследственная мозжечковая атаксия, мозжечковая гередоатаксия, наследственная мозжечковая атаксия Мари).
Наследственная болезнь, характеризующаяся медленно нарастающими мозжечковыми расстройствами в сочетании с поражением пирамидных путей. Наследуется по аутосомно-до- минантному типу. Проявляется атаксией, повышением сухожильных рефлексов, замедленной скандированной речью, глазодвигательными расстройствами и нарушениями зрения.
Описан французским невропато-
логом P. Marie (1853–1940).
Синдром Райли–Дея (синоним –
семейная вегетативная дисфункция Райли–Дея). Наследственный син-
Рис. 7.60. Мозг умершего с признаками деменции Пика
Приведено по: http://www.iqb.es/neurologia/ atlas/pick/pick01.jpg
289

дром – сочетание гиперсаливации, пониженного слёзоотделения, эритемы, психической лабильности, гипорефлексии и сниженной болевой чувствительности. Наследуется по аутосом- но-рецессивному типу.
Описан в 1968 г. американскими врачами С. М. Riley и R. L. Day.
Синдром Стерджа–Вебера–Краб- бе (синонимы – синдром Брашфил- да–Уайетта, невоидная аменция, энцефалотригеминальный ангиоматоз,
врождённая нейроэктодермальная дисплазия, синдром Краббе, нейрокутанный синдром, болезнь Стер-
джа–Вебера, болезнь Штурге–Вебера). Врождённый ангиоматоз нервной системы, проявляющийся эпилептиформными припадками и другими расстройствами центральной нервной системы (рис. 7.61), глаукомой, сосудистыми невусами по ходу ветвей тройничного нерва.
Описан английскими врачами W. A. Sturge (1850–1919) и F. P. Weber (1863– 1962), а также датским невропатологом К. Н. Krabbe (1885–1961).
Синдром Тевенера (синонимы – acropathia ulceromutilans familiaris, се-
мейная язвенно-мутилирующая акропатия, семейный акроостеолиз, яз- венно-мутилирующая деформирующая акропатия, синдром Джиаккаи,
синдром Перона–Дроке–Кулона). Трофические язвы подошвенных поверхностей стоп вследствие наследственно детерминированного поражения спинальных ганглиев, гиперкератоз дистальных отделов конечностей (чаще нижних), остеопороз плюсневых и фаланговых костей, образование язв, деформация кистей и стоп, остеолиз мелких костей. Иногда образуется так называемая «стопа слона» вследствие вторичного лимфостаза.
Описан французским невропатологом A. Thevenard (1898–1959). Синдром Терсиля. Сочетание башнеобразной формы черепа (рис. 7.62),
экзофтальма, атрофии зрительных нервов, глазодвигательных нарушений, эпилептических припадков и олигофрении, обусловленное врождённым краниостенозом.
Синдром (болезнь) Унферрихта–Лундборга (синоним – миоклонус-эпи-
лепсия). Гетерогенное наследственное заболевание, передающееся по ауто- сомно-рецессивному типу и характеризующееся прогрессирующей эпилепсией, миоклонией (быстрыми внезапными сокращениями отдельных групп мышц), атаксией и неврологическими расстройствами. Выделяют две формы заболевания – балтийскую и средиземноморскую. Генетический фактор болезни (ЕРМI) локализован на участке q22.3 хромосомы 21.
Впервые описан в 1891 г. шведским врачом Н. В. Lundborg (1868–1943).
290