

Ч 2
.pdfтип В (висцеральная, или хроническая, форма заболевания) – отличается поздним началом и распространённым поражением внутренних органов, при этом поражения нервной системы, как правило, не наблюдается;
тип С (подострая, или юношеская, форма заболевания) – характеризуется медленным прогрессированием неврологической симптоматики, гепато- и спленомегалией, анемией, судорогами, мозжечковыми расстройствами; у пациентов могут быть тонкие конечности, коричневые пятна на коже (виш- нёво-красные пятна на сетчатке глаз – пятна Пика).
Прогноз заболевания неблагоприятный.
Описан в 1914 г. немецким врачом А. Niemann (1880–1921), а затем в 1922 г. его соотечественником L. Pieck (1868–1944).
Синдром (болезнь) Негели I. Наследственная болезнь из группы геморрагических диатезов, обусловленная нарушением процессов агглютинации тромбоцитов и характеризующаяся резко увеличенным временем кровотечения и временем ретракции сгустка при нормальном времени свёртывания крови. Наследуется по аутосомно-доминантному типу.
Описана швейцарским гематологом О. Naegeli (1871–1938).
Синдром (болезнь) Норума. Врождённое заболевание, характеризующееся аномальным накоплением лецитина и неэтерифицированного холестерина в крови, почечной недостаточностью и гипохромной анемией. Наследуется по аутосомно-рецессивному типу. Обусловлено дефицитом лецитин-холестери- нацетилтрансферазы (ген LCAT локализован на участке q22.1 хромосомы 16).
Синдром (болезнь) Оккермана (синонимы – болезнь Оккермана– Кжелльмана, маннозидоз). Заболевание обусловлено отсутствием ферментов печени – маннозидаз А и В (гены MANA и MANB локализованы соответственно на участках q11–qter хромосомы 15 и cen–q13.1 хромосомы 19). Наследуется по аутосомно-рецессивному типу. В основе болезни лежит врождённое отсутствие лизосомального фермента альфа-маннозидазы в мозге, печени и селезёнке. При этом внутриклеточно накапливаются мукополисахариды с большим содержанием маннозы. Проявляется гепато- и спленомегалией, мышечной гипотонией сразу после рождения, грубой задержкой психического и двигательного развития, помутнением капсулы хрусталиков, наличием пенистых клеток в костном мозге, гипогаммаглобулинемией. Также часто отмечаются высокий рост, увеличенный язык, мышечная гипотония и различные аномалии скелета. К 4–5-му годам жизни, как правило, болезнь заканчивается смертельным исходом вследствие снижения иммунитета и развития инфекционных осложнений.
Описан в 1964 г. P. A. Ockerman и в 1969 г. L. Kjellman с соавт.
Синдром Олбрайта–Баттлера (синоним – почечный ацидоз Олбрайта).
Вариант синдрома Лайтвуда–Олбрайта, обусловленный наследственным дефектом секреции водородных ионов в дистальных отделах извитых канальцев почек. Наследуется по доминантному (сцепленному с полом) типу. Проявляется в конце 1-го – начале 2-го года жизни искривлением нижних конечностей
261
уначинающих ходить детей. Признаки заболевания напоминают рахит или остеомаляцию (задержка роста, деформация костей скелета, особенно голеней, ограничение подвижности в крупных суставах – тазобедренных, коленных, локтевых), из-за которой походка становится неуверенной, а в тяжелых случаях ребенок вообще перестаёт ходить. Характерны компенсаторный поясничный лордоз, удлинение черепа в переднезаднем направлении, болезненность костей и мышц. В отличие от витамин D-дефицитного рахита нет нарушения общего состояния, мышечной гипотонии, спазмофилии. У больных гетерозиготных женщин клинические проявления выражены меньше, чем
умужчин. Отмечают гипофосфатемию, повышение уровня щелочной фосфатазы при нормальных показателях кальция, гиперфосфатурию. При рентгенологическом исследовании имеют место рахитоподобые изменения костей скелета, грубоволокнистая структура губчатого вещества костей.
Описан в 1937 г. американскими врачами F. Albright, A. Buttler и E. Bloomberg.
Синдром Пальтауфа (синонимы – карликовость Пальтауфа, примор-
диальный нанизм). Врождённая болезнь, характеризующаяся сочетанием симптомов лимфатико-гипопластического диатеза и карликовости с сохранением нормальных пропорций тела.
Описан чешским врачом A. Paltauf (1860–1893).
Синдром (болезнь) Пелицеуса–Мерцбахера. Врождённое заболевание,
являющееся одной из форм лейкодистрофии. Передается по рецессивному (сцепленному с полом) типу, обусловлено дефектом протеолипидного белка, ген которого (PLP) локализован на участке q21.33–q22 Х-хромосомы. Форма спастического церебрального паралича, проявляющегося в первые месяцы жизни. Вызвана аплазией подкоркового белого вещества больших полушарий с демиелинизацией нервных волокон. Неврологическими симптомами заболевания являются нистагм, дрожание головы, нарушения моторики и речи, спастические парезы и параличи. В первые годы болезнь развивается с большей прогредиентностью, а затем может на долгое время стабилизироваться. Во всех случаях заболевания с относительно ранним его началом имеется задержка умственного развития, при более позднем начале отмечается органическое снижение психики, обычно не достигающее деменции.
Описан в 1885 г. врачом F. Pelizaeus и в 1910 г. врачом L. Merzbacher.
Синдром Пельгера (синонимы – аномалия лейкоцитов Пельгера, ано-
малия Пельгера–Хюэта). Наследственная аномалия, при которой ядра нейтрофилов не сегментированы, имеют форму эллипса, боба или почки, иногда с намечающейся перетяжкой посередине. Наследуется по аутосомно-доми- нантному типу.
Описана голландским врачом К. Pelger (1885–1931).
Синдром Пендреда. Наследственная болезнь, характеризующаяся нарушением биосинтеза тиреоидных гормонов и проявляющаяся врождённым гипотиреозом, узловым зобом и глухонемотой. Наследуется по аутосомно-ре- цессивному типу.
Описан английским врачом V. Pendred (1869–1946).
262

Рис. 7.38. Картина крови (анемия, нейтропения, лактоацидоз) при синдроме Пирсона Приведено по: http://path.upmc.edu/cases/case104/images/micro2.jpg
Синдром Пирсона. Наследственное заболевание, характеризующееся анемией (рис. 7.38), вакуолизацией костного мозга, аномалиями поджелудочной железы и другими симптомами. Заболевание обусловлено крупными делециями митохондриального генома и по фенотипическим проявлениям отличается от синдрома Кирнса–Сейра доминированием мутантных молекул мтДНК в различных группах поражённых тканей.
Описан американским врачом С. М. Pearson.
Синдром Прасада–Коуза (синоним – врождённая агаммаглобулинемия с лимфаденопатией). Наследственная болезнь, характеризующаяся отсутствием в крови гаммаглобулинов, проявляющаяся увеличением всех лимфатических узлов, печени, селезёнки и гемолитической анемией. Встречается у женщин.
Описан в 1962 г. американскими врачами A. S. Prasad и D. W. Koza.
Синдром Рейманна (синоним – периодическая болезнь). Наследственное заболевание, связанное с врождёнными энзимопатиями, сопровождающееся нарушением равновесия в ряде систем – протеолитической, белково-синтети- ческой, гормональной и иммунной. Наблюдается преимущественно в определённых этнических группах (у евреев, арабов, армян). Заболеванию обычно предшествует группа неспецифических симптомов (недомогание, чувство дискомфорта, раздражительность, слабость, ломота в суставах, озноб и др.), возникающих за несколько часов до острой атаки. Характеризуется периодичными (через определённые промежутки времени) приступами болей в животе или ознобами в сочетании с неукротимой рвотой, фебрильной гипертермией, гиперлейкоцитозом (абдоминальная форма заболевания). При торакальной форме болезни отмечается болевой синдром в грудной клетке, при суставной форме заболевания имеют место болевые приступы в суставах. Приступы повторяются часто на протяжении ряда лет, но не приводят к органическим из-
263

менениям со стороны внутренних органов и систем. Дифференциальную диагностику проводят с острой абдоминальной патологией, инфарктом миокарда, инфекционной патологией и заболеваниями суставов.
Описан в 1937 г. американским врачом Н. A. Reimann.
Синдром (болезнь) Рефсума (синонимы – наследственная сенсомоторная полиневропатия IV типа, наследственная полиневропатическая атак-
сия). Редкое наследственное (с аутосомно-рецессивным типом наследования) заболевание (нередко с кровным родством родителей пациентов), связанное с нарушением метаболизма и накоплением в организме фитановой кислоты. Характеризуется главным образом поражением периферической нервной системы (полиневропатия), мозжечковой атаксией, нарушением зрения (пигментная дистрофия сетчатки) и развитием ювенильного сахарного диабета. Патогенез окончательно не изучен. При биохимическом исследовании выявляется повышение содержания в крови фитановой кислоты (20-углеродной жирной кислоты с разветвлённой цепью) и её значительное накопление в периферической нервной системе, головном и спинном мозге, печени, почках, сердце и других органах, что позволяет отнести это заболевание к группе наследственных липидозов. В нервной системе обнаруживают распространённые дегенеративные изменения, локализующиеся преимущественно в нервах, их корешках, клетках передних рогов спинного мозга, мозжечковых путях и сетчатке глаза. Отмечается также утолщение мягкой мозговой оболочки. В результате поражения периферической нервной системы появляются парестезии в ногах и развивается двусторонний парез разгибателей стопы, ведущий к изменению походки. Наблюдается мозжечковая атаксия, снижается интеллект. Развитие пигментной дистрофии сетчатки и частичной атрофии зрительных нервов способствует значительному ухудшению зрения. Нередко наблюдаются также деформация стопы (полая стопа) (рис. 7.39), потеря обоняния, глухота (в результате поражения слуховых нервов), изменения кожи по типу ихтиоза и кардиомиопатия. В цереброспинальной жидкости отмечается
Рис. 7.39. Стопы пациента с синдромом Рефсума
Приведено по: http://qjmed.oxfordjournals. org/content/vol94/issue8/images/large/QJ75052.2.2.jpeg
264

белково-клеточная диссоциация. Течение заболевания медленно прогрессирующее, иногда стационарное. Болезнь может выявляться как у детей, так и у взрослых (обычно в возрасте до 40 лет).
Диагноз устанавливают на основании характерной клинической картины, данных исследования глазного дна, цереброспинальной жидкости, электромиографических признаков невропатии. Прогноз для жизни в целом благоприятный, однако при поражении сердца возможна внезапная смерть.
Описан в 1947 г. норвежским невропатологом S. B. Refsum.
Синдром Рихнера–Ханхарта (синонимы – Richner–Hanhart syndrome, tyrosinemia II). Наследственное заболевание человека, проявляющееся кератозом ладоней и подошв, а также дистрофией роговицы глаз, задержкой развития и умственной отсталостью, низкорослостью, образованием множественных липом, нарушением метаболизма тирозина (из-за дефицита тирозинаминотрансферазы). Наследуется по аутосомно-рецессивному типу (ген TAT локализован на участке q22.1 хромосомы 16).
Описан в 1938 и 1947 гг. врачами H. Richner и E. Hanhart.
Синдром Ротмунда (синонимы – синдром Ротмунда–Томсона, ретикулярная атрофическая пойкилодермия, атрофический телеангиоэктатический дерматоз с катарактой, синдром Томсона). Редкое наследственное аутосомно-рецессивное заболевание, обусловливающее развитие в раннем детском возрасте совокупности фенотипических признаков – нанизма, пойкилодермии лица и конечностей (рис. 7.40), двусторонней катаракты, дистрофии волос, ногтей, зубов и роговицы, дефицита гормона роста, нарушений эндохондрального окостенения и артериосклероза. Популяционная частота неизвестна. К настоящему времени описано более 80 случаев заболевания. Нередко наблюдается кровное родство родителей. Мужчины поражаются реже.
Заболевание часто сопровождается гипертрихозом, гипоплазией потовых и сальных желёз. Описаны аллопеция, раннее облысение, редкие брови и ресницы или их отсутствие, дистрофия ногтей. Один из кардинальных признаков синдрома – двусторонняя катаракта, которая развивается быстро, обычно в возрасте 4–8 лет (ювенильная катаракта). Среди прочих клинических симптомов можно отметить раннее поседение, дистрофию ногтей и нарушение развития зубов. У пациентов нередко развивается так называемый ювенильный артериосклероз. Из более редких поражений следует отметить акромикрию (малые размеры кистей и стоп, короткие
265
пальцы), а также ряд эндокринных нарушений – гипогенитализм, гипоплазию яичек, слабое развитие вторичных половых признаков и аменорею. При гистологическом исследовании кожи выявляются атрофия эпидермиса, истончение коллагеновых и гибель эластических волокон, а также атрофия придатков кожи. При гиперпигментации – увеличение уровня меланина в базальных клетках, при депигментации – перемещение его в дерму. При рентгенографическом исследовании костей часто обнаруживают эпи- и метафизарные нарушения энхондральной оссификации, дизостозы и дисплазии костей. Молекулярно-гене- тические исследования с использованием FISH-технологии выявляют хромосомную нестабильность, захватывающую хромосому 8 и проявляющуюся
втканевом мозаицизме по этой хромосоме. Полагают, что ген данного синдрома локализован на коротком плече хромосомы 8 (8р). Прогноз для жизни в целом благоприятный, для зрения – неблагоприятный (слепота).
Впервые описан в 1868 г. немецким дерматологом A. Rothmund и затем
в1923 г. австралийским дерматологом M. Thomson.
Синдром Руитера–Помпена. Наследуемое по рецессивному (сцепленному с Х-хромосомой) типу заболевание из группы гликозидозов, обусловленное патологическим отложением липидов в сосудах, гладких мышцах и других органах с нарушением их функций.
Впервые описан голландским дерматологом М. Ruiter и врачом A. W. Pompen.
Синдром Саньян–Сакати. Врождённое заболевание с аутосомно-рецес- сивным типом наследования, проявляющееся гипопаратирозом, ассоциированным с тяжёлой задержкой роста и дизрафическим статусом пациентов. Описан среди членов семей из западных провинций Саудовской Аравии при близкородственных браках, верифицирован среди бедуинов. Ген, ответственный за развитие синдрома, локализован на 1q42–43. С помощью молекулярногенетических исследований идентифицированы мутации гена ТВСЕ, кодирующего тубулинспецифические шаперон-белки, которые необходимы для приобретения правильной конформации (фолдинга) а-субъединиц тубулина и образования а-b-тубулин-гетеродимеров.
Синдром (болезнь) Санфилиппо (синоним – мукополисахаридоз III
типа). Является врождённым заболеванием с аутосомно-рецессивным типом наследования. Выделяют несколько типов данного синдрома – А, В, С и D, которые не отличаются клинически, но характеризуются разными биохимическими дефектами. При типе А отсутствует гепарин-N-сульфатаза, при типе В – N-ацетил-альфа-глюкозаминидаза, при типе С – ацетил-КоА-L-глюкоза- минид-N-ацетилтрансфераза, при типе D – N-ацетил-глюкозаминсульфат- сульфатаза. Заболевание проявляется накоплением гепарансульфата без дерматанили кератансульфата в возрасте 2–3 лет или в школьном возрасте. Отмечаются нарушения психики, изменения поведения в виде возбудимости, неспособности к сосредоточению, агрессивности и нарушения сна (рис. 7.41), а также рецидивирующие инфекции верхних дыхательных путей. Далее про -
266

Рис. 7.41. Дегенерация клеток головного мозга при синдроме Санфилиппо Приведено по: http://www.pasteur.fr/ icono/RAR/RAR2005/photo1_Rtg.jpg
исходит снижение интеллекта и развиваются спастическая диплегия и гепатоспленомегалия. Характерные изменения выявляются со стороны скелета – утолщение костей черепа, патологические формы поясничных позвонков. Отмечается повышенная экскреция с мочой гепарана сульфата. Пациенты редко доживают до 20 лет (смерть наступает обычно в возрасте после 10 лет).
Описан в 1954 г. американским педиатром S. Sanfilippo с соавт. Синдром Сидбери. Наследственное заболевание человека с выраженны-
ми признаками нарушения жирового обмена. Проявляется сразу после рождения в виде судорог, летаргического состояния и гепатоспленомегалии. В крови уменьшено число лейкоцитов и тромбоцитов. Моча имеет своеобразный запах, обусловленный наличием в ней масляных и гексоновых кислот. Прогноз неблагоприятный – дети погибают от сепсиса.
Описан американским педиатром J. B. J. R. Sidbury.
Синдром Симонса (синонимы – болезнь Симонса–Холландера, синдром Смита II, синдром Бэрраквера, парадоксальная липодистрофия).
Ограниченная атрофия жировой ткани (чаще верхней половины тела и лица) при сохранённой подкожной основе на других участках тела. Поражаются голень или бедро, ягодицы. Иногда определяют костные кисты, дебильность, гипертиреоз и вазомоторные расстройства. Болеют чаще женщины. Этиопатогенез не известен. Предполагается врождённый характер заболевания.
Описан в 1911 г. немецким врачом A. Simons.
Синдром Сиппла (синонимы – Sipple syndrome, множественная эндо-
кринная неоплазия IIa типа). Аутосомно-доминантно наследуемое заболевание, обусловленное генетическими дефектами. Характерны медуллярная карцинома щитовидной железы (С-клеточная карцинома, выявляемая практически у всех пациентов; как правило, многоочаговые опухоли, развитию которых предшествует гиперплазия C-клеток и которые секретируют кальцитонин,
267

реже – адренокортикотропный гормон или серотонин) или сочетание гиперплазии С-клеток, феохромоцитомы (более чем у 70 % пациентов с выраженной артериальной гипертензией) и аденомы (гиперпаратиреоидизм, выявляемый примерно у 50 % пациентов) паращитовидных желёз; возможен зудящий дерматит в межлопаточной области. Нередко сочетается с различными врождёнными дефектами.
Варианты синдрома Сиппла (обусловлены мутациями протоонкогена c-ret): а) классическая МЭН IIa типа (сочетание медуллярного рака щитовидной железы, феохромоцитомы (рис. 7.42) и гиперплазии или опухолей паращитовидных желёз; этот вариант может быть обусловлен мутациями в половых
клетках и соматическими мутациями); б) семейный изолированный медуллярный рак щитовидной железы (этот
вариант МЭН IIa типа редко включает другие опухоли; рак щитовидной железы обусловлен множественными мутациями в половых клетках; вариант характеризуется высокой пенетрантностью);
в) МЭН IIa типа с первичным амилоидозом кожи (наблюдаются краснокоричневые высыпания в виде пятен или узелков между лопатками или на голенях, сопровождающиеся мучительным зудом);
г) МЭН IIa типа с болезнью Гиршспрунга; у 93–95 % пациентов обнаруживают точечные мутации протоонкогена c-ret (10q11), который кодирует ре-
Рис. 7.42. Феохромоцитома правого надпочечника у пациента с синдромом Сиппла (данные компьютерной томограммы)
Приведено по: http://home.earthlink.net/~radiologist/tf/012201a.jpg
268
цептор нейротропного фактора, регулирующего пролиферацию и дифференцировку клеток – производных нервного гребня; мутации c-ret приводят к активации тирозинкиназы рецептора и трансформации нейроэктодермальных клеток и могут затрагивать разные кодоны c-ret (так, у всех больных классической МЭН IIa типа и МЭН IIa типа с первичным амилоидозом кожи имеются мутации в кодоне 634, а при сопутствующей болезни Гиршспрунга – мута-
ции в кодонах 609, 618, 620 и 634).
Расшифровка генетических дефектов, лежащих в основе синдрома, позволяет оценивать риск заболевания у родственников пациентов и планировать профилактические мероприятия.
Описан в 1961 г. врачом J. H. Sipple.
Синдром Спренгера (синоним – синдром муколипидоза I типа). В осно-
ве заболевания лежит наследственный дефицит (или полное отсутствие) бета- N-ацетилнейраминидазы в лейкоцитах и фибробластах. Клинические проявления болезни отмечаются в конце первого года жизни отставанием в психомоторном развитии. На глазном дне обнаруживаются проявления типа «вишнёвой косточки», в некоторых случаях имеет место помутнение роговицы. Неврологические проявления, которые манифестируют в возрасте старше 4 лет, проявляются мышечной гипотрофией и гипотонией, атаксией, судорожным синдромом, тремором и признаками периферической нейропатии. Выявляются гепатомегалия, спленомегалия и грыжи живота.
Описан в 1968 г. J. Spranger.
Синдром Тигсгрупа. Наследственное заболевание, характеризующееся нарушением выделения связанного билирубина из печёночных клеток и мелких желчных протоков. Клинически проявляется интермитирующей желтухой с высоким уровнем прямого билирубина в крови, кожным зудом и гепатоспленомегалией.
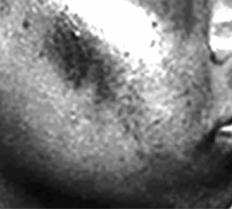
Синдром Труазье–Ано–Шоффара (синонимы – пигментный цирроз печени, сидерофилия, бронзовый диабет, гемохроматоз). Общее заболевание,
характеризующееся нарушением обмена железа, повышенным его содержанием в сыворотке крови и накоплением в тканях и внутренних органах. Встречается сравнительно редко, преимущественно у мужчин.
Первичный («идиопатический») гемохроматоз относят к ферментопатиям и рассматривают как врождённое заболевание с аутосомно-доминантным типом наследования, при котором в тонком кишечнике человека всасывается значительно больше железа, чем в норме, и оно избыточно депонируется (в основном в виде нерастворимого гемосидорина) в тканях – в печени, поджелудочной железе, сердечной мышце, коже и т. д. В результате возникает вторичная воспалительно-склеротическая реакция тканей поражённых органов. При этом в гладких мышцах избыточно откладывается липофусцин, в эпидермисе – меланин.
Вторичный гемохроматоз возникает при избыточном поступлении в организм железа при длительном неконтролируемом лечении препаратами железа,
269
|
при частых переливаниях крови и т. д. |
|
|
Для синдрома характерны гиперпиг- |
|
|
ментация кожи (особенно открытых |
|
|
частей тела, подмышечных впадин, ла- |
|
|
доней, половых органов и старых кож- |
|
|
ных рубцов), принимающей серо-бу- |
|
|
рый или коричневатый цвет (рис. 7.43); |
|
|
увеличенная и плотная печень; сахар- |
|
|
ный диабет; нередки явления недоста- |
|
|
точности эндокринных желёз (в пер- |
|
|
вую очередь половых). Гемосидероз |
|
|
миокарда проявляется электрокардио- |
|
Рис. 7.43. Гиперпигментация кожи лица |
графическими признаками дистрофии |
|
миокарда, аритмиями, сердечной недо- |
||
при синдроме Труазье–Ано–Шоффара |
||
Приведено по: http://www.ellensplacesalon. |
статочностью. В сыворотке крови по- |
|
com/hyperpigmentation.jpg |
вышена концентрация железа (до 50– |
|
|
70 мкмоль/л), насыщенность железом |
трансферрина достигает 90 % (в норме 30 %). Гиперферремия выявляется ещё до того, как возникают гиперпигментация кожи и увеличение печени. Исследование окрашенных голубым нитропруссидом гистологических препаратов, приготовленных из биоптатов печени пациента, позволяет обнаружить большое количество внутриклеточного железа. Отмечается повышенное выделение железа с мочой после инъекции дефероксамина. Биохимические сдвиги, характерные для цирроза печени, появляются в более позднем (по сравнению с гиперпигментацией и увеличением печени) периоде заболевания. Как правило, выявляется гипергликемия и глюкозурия.
Течение заболевания постепенно прогрессирующее. Прогноз без лечения неблагоприятный. У пожилых пациентов с длительным течением заболевания в 15–20 % случаев развивается рак печени. Средняя продолжительность жизни пациентов после установления диагноза (при отсутствии лечения) не превышает 4–5 лет. Причиной гибели пациентов могут быть печёночная или диабетическая кома, острое кровотечение из варикозно-расширенных вен пищевода и желудка, сердечная недостаточность.
Описан французскими врачами Ch. Е. Troisier (1844–1919), V. Ch. Hanot (1844–1896) и А. М. E. Chauffard (1855–1932).
Синдром (болезнь) Урбаха–Вите (синоним – наследственно детермини-
рованный липоидный протеиноз). Наследуемое по рецессивному типу заболевание человека, характеризующееся поражениями кожи и слизистых оболочек, обусловленными нарушением липидного и белкового обмена. В частности, происходит аномальное отложение нерасщеплённого белка (парапротеина).
Описан американским дерматологом Е. Urbach (1893–1946) и австрийским оториноларингологом С. Wiethe (1888–1949).
270