

Ч 2
.pdfОписан в 1956 г. немецким педиатром А. Wiskott и американским педиа-
тром R. A. Aldrich.
Синдром Джоба (синонимы – рецидивирующая инфекция с гипериммуноглобулинемией Е, синдром Иоба, синдром рецидивирующих инфекций, врождённая IgE-гипериммуноглобулинемия). Врождённое заболева-
ние, проявляющееся аномально сниженным хемотаксисом нейтрофилов и моноцитов на фоне гиперпродукции IgE. Клинически у пациентов при этом рецидивируют инфекции, вызванные золотистым стафилококком. Пациенты обычно имеют характерные черты лица, нередко – рыжий цвет волос. У них выявляются «холодные» абсцессы кожи (подкожной клетчатки и мышц), дерматит, целлюлит, заметно повышенный уровень в сыворотке IgE, в том числе уровень антистафилококкового IgE, низкий уровень антистафилококкового IgA или его отсутствие. Отмечаются умеренно выраженная эозинофилия и недостаточность функции Т-супрессорных лимфоцитов.
В течение длительного времени считалось, что «холодный» абсцесс отражает дефект хемотаксиса, когда некоторые фагоциты слишком поздно достигают очага воспаления, что, возможно, объясняется действием вторичного по отношению к лимфоцитам фактора ингибиции хемотаксиса. Вместе с тем, согласно новым представлениям, дефект хемотаксиса у этих пациентов вариабелен, а основой для снижения защитной функции организма является целый комплекс факторов и их неадекватность.
Описан в 1966 г.
Синдром Ди Джорджи (синоним – синдром Ди Георги). Изолированный наследственный Т-клеточный иммунодефицит, характеризующийся триадой ведущих клинических проявлений: гипоплазией тимуса и/или паращитовидных желёз, врождённым пороком сердца. В основе заболевания лежит порок развития третьего-четвёртого глоточных карманов, возникающий между 6-й
и10-й неделями гестации, который вызывает агенезию или дисгенезию паращитовидных желёз и тимуса. Вовлечение первого и второго жаберных карманов приводит к пороку развития лицевых структур, а поражение пятого кармана проявляется широким спектром врождённых пороков сердца (как правило, с вовлечением дуги аорты). Для большинства пациентов характерны диспластические черты лица, основными их которых являются гипертелоризм, широкая переносица, «рыбий рот», антимонголоидный разрез глаз, а также диспластичные ушные раковины. У некоторых детей наблюдают
иболее грубые аномалии, такие как микрогнатия и незаращение твёрдого
имягкого нёба (рис. 4.22). Гипокальциемия различной степени тяжести и отсутствие тени вилочковой железы при рентгенографии грудной клетки относятся к частым проявлениям заболевания. Гипокальциемические судороги возникают обычно с первых дней жизни. У всех пациентов отмечается задержка умственного развития. Врождённые пороки сердца и магистральных сосудов также относятся к наиболее характерным и тяжёлым признакам заболевания. Лабораторно количественные показатели Т-клеток варьируются от
151
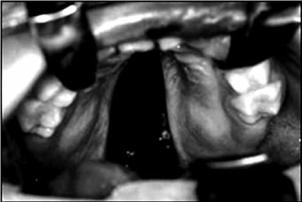
Рис. 4.22. Врождённая расщелина мягкого и твёрдого нёба у пациента с синдромом Ди Джорджи
Приведено по: http://stomatolog.md/article.php?aid=199
нормы до глубокой депрессии. Характерны диссоциация между сниженными уровнями Т- и NK-клеток и повышенным содержанием В-лимфоцитов, а также нормальные или повышенные уровни антител.
Описан в 1966 г. американским педиатром А. М. Di George.
Синдром (болезнь) Дюбуа (синоним – абсцесс Дюбуа). Врождённое за-
болевание, характеризующееся множественными кистами (величиной с го - рошину) вилочковой железы. Стенки их гладкие, полость выполнена детритом, напоминающим гной. В ряде случаев может проявляться признаками миастении.
Описан французским врачом Р. Dubois (1795–1871).
Синдром Зупаноса–Зегессера–Шнейдера. Опухоль вилочковой железы,
сопровождающаяся выраженной миастенией, эритробластической анемией, присутствием волчаночных клеток в крови (рис. 4.23).
Описан в 1970 г. французскими врачами G. Zoupanos, F. Saegesser
и С. Schneider.
Синдром (болезнь) Кавасаки (синоним – слизисто-кожно-лимфоузло-
вой синдром). Острое лихорадочное заболевание, ассоциированное с васкулитом. Этиология не известна. Предполагается аутоиммунный характер поражения. Болеют преимущественно дети до 11 лет.
Для заболевания характерны симптомы асептического менингита, диарея, артралгии или артриты, миокардит и коронарный васкулит. В острую фазу возможно развитие панкардита. У некоторых детей прогрессирование коронарного васкулита приводит к некрозу сосудистой стенки, образованию аневризм и тромбозу. Факторы риска развития коронарных аневризм – мужской пол, начало болезни до 18 мес., японское происхождение, острое токсическое лихорадочное течение с ранней картиной миокардита.
152

Рис. 4.23. Опухоль тимуса с явлениями миастении, характерная для синдрома Зупаноса–Зе- гессера–Шнейдера (данные компьютерной томографии)
Приведено по: http://www.stormed.ru/diseases/neuro/myastenia.php
Причинами смерти являются инфаркт миокарда, аритмии, разрыв аневризм коронарных артерий. В диагностике помогает раннее выявление тромбоцитоза, повышения острофазовых показателей воспаления, лейкоцитоза с нейтрофильным сдвигом, протеинурии.
Описан японским педиатром Т. Kawasaki.
Синдром Костманна (синоним – агранулоцитоз новорождённых). На-
следственное заболевание с аутосомно-рецессивным типом наследования. Клинически проявляется тяжёлыми рецидивирующими гнойными инфекционными процессами различной локализации с первых месяцев жизни. Имеют место сочетанные проявления – например, замедление умственного развития, микроцефалия, катаракта (рис. 4.24) и низкорослость. Костный мозг миелоклеточный, без миелоидных и лимфоидных элементов, в нём определяется миелоидная гипоплазия на стадии промиелоцита. Для гематологической картины заболевания характерна триада симптомов – врождённый агранулоцитоз, лимфопения, отсутствие клеточного и гуморального иммунитета. У младенцев развиваются тяжёлые рецидивирующие гнойные инфекции с первых месяцев жизни. Лимфатические узлы, миндалины, тимус отсутствуют, иногда имеют место анемия и тромбоцитопения.
Описан в 1954 г. шведским педиа-
тром R. Kostmann.
Синдром Луи-Бара (синоним –
синдром атаксии-телеангиэктазии). |
Рис. 4.24. Врождённая катаракта, характер- |
Первичный (врождённый) иммуноде- |
ная для синдрома Костманна |
фицит, обусловленный дефектом со- |
Приведено по: http://mntk.irkutsk.ru/disease. |
зревания, снижением функции Т-лим- |
php?idp=3 |
153
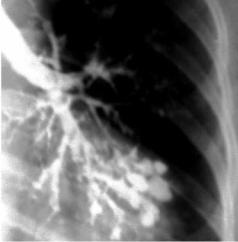
фоцитов, уменьшением их числа в крови (особенно Т-хелперов), дефицитом иммуноглобулинов (в первую очередь IgA, IgE, реже – IgG). Синдром характеризуется сочетанием атаксии и других неврологических отклонений с телеангиэктатическими изменениями сосудов склер, лица. Поражение нервной системы проявляется симптомами выпадения функций мозжечка, подкорковых ганглиев, диэнцефальной области, пирамидной системы. В результате их поражения отмечаются нарушение походки, замедленность произвольных движений, гиперкинезы, вегетососудистая дистония. У многих пациентов диагностируется вялотекущая пневмония, развиваются ателектазы, пневмосклероз и бронхоэктазы
(рис. 4.25). При исследовании лимфатической системы устанавливают гипоплазию вилочковой железы, лимфатических узлов, селезёнки. У многих детей отмечается уменьшение содержания в крови лимфоцитов, снижена реакция бласттрансформации лимфоцитов, не определяется IgA. Заболевание характеризуется аутосомно-рецессивным типом наследования.
Прогноз синдрома неблагоприятен. Около 50 % летальных исходов обусловлено хроническим поражением бронхолёгочной системы, около 20 % – развитием злокачественных процессов, которые связывают с утратой функциональной активности тимус-зависимых лимфоцитов, а в общем плане – с отсутствием цензорной функции вилочковой железы (функции иммунологического надзора). Некоторые пациенты доживают до 40–50 лет.
Описан в 1941 г. французским врачом D. Louis-Bar.
Синдром Незелофа (синонимы – алимфоцитоз Незелофа, лимфоци-
тарная дисгенезия). Наследственно обусловленная недостаточность клеточных механизмов специфической защиты против инфекции. В патогенезе основную роль играет гипоплазия вилочковой железы, в которой, по данным гистологического исследования, отсутствуют лимфоидные клетки и тельца Гассаля. В периферической крови значительно уменьшено количество лимфоцитов.
Заболевание проявляется в первые месяцы жизни упорным септическим процессом с септико-пиемическими очагами в лёгких, среднем ухе, толстой кишке, почках и мочевыводящих путях, а также в коже. В перифе - рической крови отмечается крайне низкое содержание лимфоцитов, резко угнетена реакция бласттрансформации лимфоцитов. Слабо выражена ре-
154
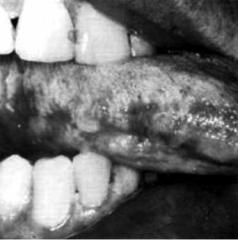
плакия слизистых оболочек (рис. 4.26); дистрофия ногтей. На втором десятилетии жизни у пациентов появляется нейтропения или апластическая анемия.
Впервые описан в 1951 г. немецким врачом F. Zinsser (1865–1952), амери-
канскими дерматологами Н. N. Cole и М. F. Engman (1869–1953).
Синдром Черджа–Строса (синоним – аллегически-гранулематозный ангиит). Заболевание аутоиммунной природы, основными проявлениями которого являются аллергическая бронхиальная астма, эозинофилия и образование лёгочных инфильтратов. В основе заболевания лежит распространённый некротический васкулит, поражающий сосуды в лёгких, нервах, коже, почках, пищеварительном тракте.
Клинически проявляется болями в животе, поносами, перфорациями кишечника, язвами гастродуоденальной зоны (нередко – с геморрагическим синдромом), псевдополипами толстой кишки с хроническим язвенным колитом, формированием гранулём в желудке, печени, почках, сальнике. В отличие от узелкового периартериита проявляется наличием экстравазальных гранулём в сочетании с эозинофильными инфильтратами в тканях и некротическим васкулитом.
Описан в 1957 г. американскими патологами J. Churg и L. Strauss.
Синдром Чедиака–Xигаши (синоним – синдром Чедиака–Хигаси). Ред-
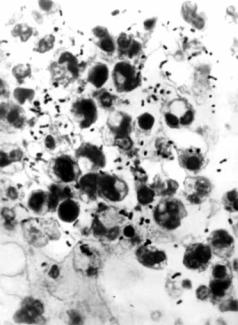
ко встречающаяся врождённая аномалия с аутосомно-рецессивным типом наследования. Клинически проявляется рецидивирующей пиогенной инфекцией, периодонтитом, частичным кожно-глазным альбинизмом (обусловленным дефектом развития лизосом и меланоцитов), нистагмом и прогрессирующей периферической нейропатией. Характерны наличие гигантских цитоплазматических гранул в лейкоцитах, тяжёлая гранулоцитопения. Почти у 50 % пациентов в пубертатном периоде развивается лимфоматозная (ускоренная) фаза заболевания. Нейтрофилы и другие типы клеток, содержащие лизосомы, имеют крупные гранулы. Отмечаемое при данном синдроме нарушение хемотаксиса нейтрофилов и моноцитов и снижение интенсивности уничтожения микроорганизмов обусловлено замедленным слиянием лизосомных гранул с фагосомами.
Описан кубинским врачом А. М. Chediak и японским врачом О. Higashi.
Синдром Швахмана–Даймонда–Оски. Врождённое заболевание с ауто-
сомно-рецессивным типом наследования в виде нейтропении, сочетающейся с недостаточностью функции поджелудочной железы. Характерны повторные инфекции со стеатореей в первые годы жизни. Стеаторея может быть скрытой и определяться только при исследовании кала на содержание жира. Отмечаются сочетанные проявления – низкорослость (60 %), задержка умственного развития (15 %), метафизарный дизостоз (30 %). Костный мозг с нормальной или уменьшенной клеточностью и миелоидным созреванием (возможна гиперплазия эритроидного ростка). Данная нейтропения поддаётся терапии колониестимулирующими факторами.
Впервые описан в 1951 г. американскими врачами Н. Shwachman
и L. K. Diamond.
156
англ. to help – помогать) содружественно оказывает помощь В-лимфоцитам в продукции антител, а субпопуляция Т-лимфоцитов-супрессоров (от англ. to suppress – подавлять) подавляет продукцию антител работающими В-лим- фоцитами по механизму обратной связи.
Открыт в 1968 г. австралийскими учёными J. Miller и J. Mitchell.


Глава 5
ВІДЫ ХИРУРГИЧЕСКОЙ ИНФЕКЦИИ
«...Каждая хирургическая операция представляет эксперимент бактериологии».
L. Moynihan (1920)
Классификация заболеваний, которые в соответствии с МКБ-10 можно отнести к хирургическим инфекциям (прямо или опосредованно, за счёт инфекционных осложнений, вызванных этими процессами).
Класс I. Некоторые инфекционные и паразитарные болезни (A00–B99). Кишечные инфекции (A00–A09):
A01 – тиф и паратиф;
A02 – другие сальмонеллёзные инфекции;
A06 – амебиаз;
A07 – другие протозойные кишечные болезни;
A08 – вирусные и другие уточнённые кишечные инфекции;
A09 – диарея и гастроэнтерит предположительно инфекционного происхождения (при отсутствии уточнения могут рассматриваться как болезни неинфекционного происхождения, указанные состояния должны классифицироваться в рубрике K52.9).
Туберкулёз (A15–A19):
A15 – туберкулёз органов дыхания, подтверждённый бактериологически и гистологически;
A16 – туберкулёз органов дыхания, не подтверждённый бактериологически или гистологически;
A17 – туберкулёз нервной системы;
A18 – туберкулёз других органов;
A19 – милиарный туберкулёз.
Некоторые бактериальные зоонозы (A20–A28):
A20 – чума;
A21 – туляремия;
A22 – сибирская язва;
A23 – бруцеллёз;
A24 – сап и мелиоидоз;
A25 – лихорадка от укуса крыс;
A26 – эризипелоид;
A27 – лептоспироз;
A28 – другие бактериальные зоонозы, не классифицированные в других рубриках.
160