

Ghai Essential Pediatrics8th
.pdfFluid and Electrolyte Disturbances -
|
|
|
|
|
Assessment of volume status |
|
|
|
|
|
|
|
|
|||
|
|
Hypovolemia |
|
|
=i |
l |
|
|
Hypervolemia |
|
||||||
|
|
|
|
|
Euvolemia |
|
|
|
||||||||
• |
Total body water .J, |
|
|
Total body water |
• |
Total body water |
i |
|||||||||
|
J - |
|
|
+ |
|
|
J |
|
|
|||||||
|
|
Total body sodium U |
|
Total body sodiumiH |
|
|
|
Total body sodiumii |
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
• |
|
r- ! |
|
f |
|
• |
|
.--t- |
||||||||
|
|
• |
|
|
|
|
|
-. |
|
|||||||
U [Na] >20 |
U [Na] <20 |
U [Na•] >20 |
|
U [Na] >20 |
|
|
u [Na] <20 |
|||||||||
Renal losses |
|
Extrarenal |
SIADH |
|
Acute, chronic |
Nephrotic syndrome |
||||||||||
Diuretics |
|
losses |
Glucocorticoid deficiency |
renal failure |
Cirrhosis |
|||||||||||
Mineralocorticoid deficiency |
Vomiting |
Hypothyroidism |
|
|
|
|
Congestive heart failure |
|||||||||
Salt losing nephropathy |
Diarrhea |
Stress |
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
||||||||
Cerebral salt wasting |
Third spacing |
Drugs |
|
|
|
|
|
|
|
|
|
|||||
Renal tubular acidosis |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
Fig. 5.5: Diagnostic approach to hyponatremia. U(Na•) urinary sodium, mEq/1; i increased; !decreased
slower rate. For symptomatic patients a combination of intravenous infusion of hypertonic saline (3% sodium chloride) and fluid restriction rapidly increase serum sodium by 5-6 mEq/1 and ameliorates the symptoms. Thereafter, if the symptoms remit, the remaining deficit can be corrected slowly over the next 1 to 3 days. When slow correction of hyponatremia in a volume-expanded patient is indicated, water restriction alone, or if this is unsuccessful, in combination with a loop-diuretic such as furosemide is preferred. Serum sodium concentration should be monitored every 2 to 4 hr and appropriate adjustments made.
Aggressive therapy with hypertonic saline in patients with chronic hyponatremia (where brain adaptation to hypo-osmolality has set in by extrusion of intracellular electrolytes and organic osmoles) can lead to osmotic demyelination called central pontine myelinolysis. This condition, which isoften irreversible is reportedinpatients with liver disease, severe malnutrition and hypoxia. Patients generally become symptomatic 2 to 7 days followingrapidcorrection (>25mEq/1inthe first24-48 hr) of chronic hyponatremia. Clinical features include mutism, dysarthria, spastic quadriplegia, ataxia, pseudo bulbar palsy, altered mental status, seizures and hypotension.
Fluid restriction alone has no role in the management of symptomatic hyponatremia. Normal saline is also inappropriate for treating hyponatremic encephalopathy due to non-hemodynamic states of vasopressin excess, such as SIADH and postoperative hyponatremia, as it is not sufficiently hypertonic toinduce reduction in cerebral edema. In presence of elevated ADH levels there is impaired ability to excrete free water with the urine osmolality exceeding that of plasma. V2-receptor anta gonists or vaptans that block the binding of ADH to its V2 receptor, are yet not recommended for treatment of hyponatremic encephalopathy. These agents may have a role in treating euvolemic hyponatremia from SIADH and hypervolemic hyponatremia in congestive heart failure.
Hypernatremia
Hypernatremia is defined as increase in serum sodium concentration to levels more than 150 mEq/1. It may be accompanied by the presence of low, normal or high total body sodium content. The major cause of hypernatremia is loss of body water, inadequate intake of water, a lack of antidiuretic hormone (ADH), or excessive intake of sodium (e.g. solutions with high sodium such as sodium bicarbonate) (Table 5.6).Diabetesinsipidusmayresultfrom a deficiency of ADH or its end organ unresponsiveness.
In the presence of an intact thirst mechanism, a slight increase in serum sodium concentration (3 to 4 mEq/1) above normal elicits intense thirst. The lack of thirst in the presence of hypernatremia in a mentally alert child indicates adefectin eithertheosmoreceptors orthe cortical thirst center. The most objective sign of hypernatremia is lethargy or mentalstatuschanges,whichproceedsto coma and convulsions. With acute and severe hypernatremia, the osmotic shift of water from neurons leads to shrinkage of the brain and tearing of the meningeal vessels and intracranialhemorrhage;slowlydevelopinghypematremia
Table 5.6: Causes of hypernatremia Net water loss
Pure water loss
Insensible losses Diabetes insipidus Inadequate breastfeeding
Hypotonic fluid loss
Renal: Loop, osmotic diuretics,postobstructive,polyuric phase of acute tubular necrosis
Gastrointestinal: Vomiting, nasogastric drainage, diarrhea; lactulose
Hypertonic sodium gain
Excess sodium intake
Sodium bicarbonate, saline infusion Hypertonic feeds, boiled skimmed milk Ingestion of sodium chloride Hypertonic dialysis
Endocrine: Primary hyperaldosteronism, Cushing syndrome

E |
_s_s_e_n_ti_a _i _P_e _d_ia_t_ri_cs----------------------- |
--------- |
|
is generally well tolerated. The latter adaptation occurs initially by movement of electrolytes into cells and later by intracellular generation of organic osmolytes, which counter plasma hyperosmolarity.
Treatment
Treatment involves restoring normal osmolality and volume and removal of excess sodium through the administration of diuretics and hypotonic crystalloid solutions. The speed of correction depends on the rate of development of hypernatrernia and associated symptoms (Box 5.2). Because chronic hypernatrernia is well tolerated, rapid correction offers no advantage and may be harmful since it may result in brain edema. Usually a maximum of 10% of the serum sodiumconcentration orabout0.5 mEq/ 1/hr should be the goal rate of correction. Seizures due to hypernatremia are treated using 5-6 ml/kg infusion of 3% saline over 1-2 hr.
POTASSIUM ------------------
Physiology
Potassium being a predominantly intracellular cation, its blood levels are unsatisfactory indicator of total body stores. Normal serum concentration of potassium ranges between 3.5 and 5 mEq/1. Common potassium-rich foods include meats, beans, fruits andpotatoes. Gastrointestinal absorption is complete and potassium homeostasis is
Box 5.2: Treatment of hypernatrernia
•Treat hypotension first, regardless of serum sodium (normal saline bolus, Ringer lactate, 5% albumin)
•Correct deficit over 48 to 72 hr. Rapid decline of chronic (>48 hr) hypematremia is associated with risk of cerebral edema. Recommended rate of drop in serum sodium is 0.5 mEq/1/hr (10-12 mEq/1/day)
•Oral solutions preferred to parenteral correction
•Generally hypotonic infusates are used (infusate sodium of - 40 mEq/1, as N/4 or N/5 saline). Sodium free fluids should be avoided (except in acute onset hypematremia, e.g. sodium overload)
•Decline of serum Na+ can be estimated using Adrogue Madias Formula:
+ |
Jinf |
+ |
(K |
+ |
Jinf |
- [ |
+ |
L) |
||
+ = {[Na |
|
|
|
Na |
||||||
D[ |
|
|
|
|
|
|
|
|
|
|
Na J |
(TBW + 1) |
|
|
|
||||||
Where [Na+] expected change in serum sodium; [Na+]inf |
||||||||||
sodium and [K+]infpotassium in 1 liter of the infusate, [Na+Js |
||||||||||
serum sodium; TBW or |
total body water = 0.6 x body |
|||||||||
weight
•Seizures due to hypematremia are treated using hypertonic (3%) saline at 5-6 ml/kg infusion over 1-2 hr.
•Renal replacement therapy (peritoneal or hemodialysis, hemofiltration) is indicated for significant hypematremia (>180-200 mEq/1) with concurrent renal failure and/or
volume overload.
• Ensure correction of ongoing fluid losses; frequent bio chemical and clinical reassessment is needed.
maintainedpredominantlythroughtheregulationof renal excretion. The fractional excretion of potassium is about 10%,chieflyregulatedbyaldosteroneat thecollectingduct. Renal adaptive mechanisms maintain potassium homeostasisuntil theglomerularfiltration ratedropstoless than 15-20 ml/min. Excretion is increasedbyaldosterone, high sodium deliveryto the collecting duct (e.g. diuretics), urine flow (e.g. osmotic diuresis), blood potassium level, glucocorticoids, ADH and delivery of negatively charged ionstothe collectingduct (e.g.bicarbonate). Inrenalfailure, the proportion of potassium excreted through the gut increases, chiefly by the colon in exchange for luminal sodium.
Aldosterone and insulin are two play important roles inpotassiumhomeostasis. Insulin stimulatedbypotassium ingestion increases uptake of potassium in muscle cells, through increased activity of the sodium pump. High potassium levels stimulate its renal secretion via aldos terone-mediated enhancement of distal expression of secretory potassium channels (ROMK). Insulin, beta adrenergic stimuli and alkalosis enhance potassium entry into cells. The reverse happens with glucagon, a-adrener gic stimuli and acidosis.
Hypokalemia
Hypokalernia is defined as a serumpotassium level below 3.5 mEq/1. The primary pathogenetic mechanisms result ing in hypokalemia include increased losses, decreased intake or transcellular shift (Table 5.7). Vomiting, a
Table 5.7: Causes of hypokalernia
Increased losses
Renal
Renal tubular acidosis (proximal or distal)
Drugs (loop and thiazide diuretics, amphotericin B, aminoglycosides, corticosteroids)
Cystic fibrosis
Gitelman syndrome, Bartter syndrome, Liddle syndrome Ureterosigmoidostomy
Mineralocorticoid excess (Cushing syndrome, hyperal dosteronism, congenital adrenal hyperplasia (11 P-hydroxy lase, 17 o:-hydroxylase deficiency)
High renin conditions (renin secreting tumors, renal artery stenosis)
Extrarenal
Diarrhea, vomiting, nasogastric suction, sweating Potassium binding resins (sodium polystyrene sulfonate)
Decreased intake or stores
Malnutrition, anorexia nervosa Potassium-poor parenteral nutrition
Intracellular shift
Alkalosis, high insulin state, medications (P2-adrenergic agonists, theophylline, barium, hydroxychloroquine), refeeding syndrome, hypokalemic periodic paralysis, malignant hyperthermia, thyrotoxic periodic paralysis

Fluid and Electrolyte Disturbances -
common cause of hypokalemia, produces volume deple tion and metabolic alkalosis. Volume depletion leads to secondary hyperaldosteronism, which enhances sodium resorptionandpotassiumsecretion inthecortical collecting tubules. Metabolic alkalosis also increases potassium secretionduetothe decreased availabilityofhydrogenions for secretion in response to sodium resorption.
Regardless of the cause, hypokalemia produces similar signs and symptoms. Symptoms are nonspecific and predominantlyare related to muscular or cardiac function.
Severe hypokalemia (<2.5 mEq/1) may cause muscle weakness (neck flop, abdominal distension, ileus) and produce cardiac arrhythmias. Chronic hypokalemia is associated with interstitial renal disease of uncertain pathogenesis. Hypokalemia increases the risk of digoxin toxicityby promotingitsbinding to myocytes, potentiating its action and decreasing clearance.
The transtubular potassium gradient (TIKG) accounts for the confounding effect of urine concentration on interpretation of urine potassium excretion.
(urine potassium x serum osmolality) TTKG = ,, . .
\.serum potassium x urme osmola11ty. )
This test cannot be applied when the urine osmolality is less than the serum osmolality. TIKG less than 4 sug gests that the kidney is not wasting excessive potassium, while a value greater than or equal to 4 indicates signifi cant renal loss.
Treatment
Patients should be evaluated to determine the underlying causes and determine whether it is associated with hypertensionandacidosisoralkalosis (Fig.5.6). Hypertension may be a clue to primary hyperaldosteronism, renal artery stenosis, or the rarer forms of genetically inherited hyper-
tension such as congenital adrenal hyperplasia, gluco corticoid remediable hypertension or Liddle syndrome.
Relative hypotension and alkalosis suggests diuretic use, or a tubular disorder such as Bartter or Gitelman syndrome.
Therapy involves decreasing ongoing losses (e.g. discontinuation of diuretics, -agonists), replenishing potassium stores (oral or intravenous administration of potassium chloride) and disease-specific therapy for the conditions such as Bartter and Gitelman syndrome (e.g. indomethacin, angiotensin-converting enzyme inhibitors) (Box 5.3). Potassium is generally replaced orally at a dose of 2-4 mEq/kg/day in 3 or 4 divided doses. Intravenous
Box 5.3: Treatment of hypokalemia
•Deficit is corrected over 24 hr
•Identify and stop ongoing losses of potassium: use of potassium sparing diuretics, intravascular volume restoration with normal saline, correction of coexisting hypomagnesemia
•Take ongoing potassium losses into consideration while replacing deficit, bymeasuring thevolume and potassium concentration of body fluid losses
•Replace the deficit: Oral route is safer than IV. Dose is 2-4 mEq/kg/day (maximum 120-240 mEq/day) in 3 or 4 divided doses. Liquid preparations are bitter and may be diluted with juice or water. Potassium chloride preferred; potassium citrate or acetate in concurrent acidosis
•IV correction is used cautiously preferably under ECG monitoring in patients unable to take oral preparation, severe hypokalemia (5 2.5 mEq/1), or associated arrhy thmias. For rapid correction: 0.5 to 1.0 mEq/kg (maximum 40 mEq) over 1 hr. Infusate potassium should not exceed 40-60 mEq/1
•Disease specific therapy for specific conditions
Hypokalemia
(serum K• <3.5 mEq/L)
Exclude spurious values
(e.g. cellular uptake in leukocytosis)
- - |
r |
* |
|
|
J |
|
|
|||||||
|
...------------===-Je-1 |
|
||||||||||||
Urine K• <20 mEq/L, TTKG <4l |
Spot urine potassium, ;e ,.... |
Urine K• |
mEq/L, TTKG |
>41 |
||||||||||
|
(nonrenal cause) |
|
urine osmolality, TTKG |
j |
( >20 |
|
|
|
||||||
|
|
|
nal K .I s ) |
|
||||||||||
|
|
·------, |
rMetabolic acidosis |
|
@etabolic alkalosisI |
|
|
|||||||
|
Gastrointestinal losses |
|
|
|
|
|
|
|
|
|
|
|
||
|
Diarrhea, small intestine |
|
|
|
|
|
|
|
|
|
|
|
||
|
drainage, villous adenoma |
|
|
|
|
|
|
|
|
|
|
|
||
|
Transcellular shift |
|
|
|
|
|
|
|
|
|
|
|
||
|
Insulin, -adrenergic agonist, |
|
|
|
|
|
|
|
|
|
|
|
||
alkalosis, periodic paralysis |
RTA type 1, 2 |
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
||||||||
|
Poor intake |
|
|
Diabetic ketoacidosis |
|
|
|
|
|
|
||||
|
Anorexia nervosa |
|
Ureterosigmoid diversion |
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Fig. 5.6: Diagnostic approach to hypokalemia. K+ potassium; RTA renal tubular acidosis; TIKG transtubular potassium gradient

E s s e n tia1 P e d i atric s |
______________________ |
|
___ |
_ _ _ _ _ _ _ _ _ _ _ _ _- ____________ |
|
|
|
|
potassium chloride may be used for correction in cases unable to take orally or those with severe hypokalernia ( 2.5 mEq/1) or associated arrhythmias.
Hyperkalemia
Hyperkalemia, defined as serum potassium level exceeding 5.5 mEq/1, is most commonly associated with renal insufficiency, acidosis and diseases that involve defects in mineralocorticoid, aldosterone and insulin function. Sudden and rapid onset of hyperkalemia is one of the most serious electrolyte disturbances and result in severe cardiac arrhythmia.
Factitious or pseudohyperkalernia can occur because of the practice of squeezing of extremities during phle botomy or blood sampled from a limb being infused with potassium-containing fluid or hemolysis of a standing sample. Thrombocytosis and leukocytosis can also lead to false elevation of serum potassium levels. True hyper kalemia is caused by one or more of 3 mechanisms: increased potassium intake, extracellular potassium shifts or decreased excretion (Table 5.8). Increased potassium intake may result from inappropriate intravenous or oral potassium supplementation. Packed red blood cells have high concentrations of potassium that can lead to hyperkalernia. Acidosis results in transcellular potassium shift, but any cellular injury that disrupts the cell mem brane (e.g. tumor lysis syndrome, rhabdomyolysis, crush injury, massive hemolysis) can cause hyperkalemia.
Patients may report nausea, vomiting and paresthesias or nonspecific findings of muscle weakness (skeletal, respiratory), fatigue and ileus. Clinical manifestations are related to the effects of elevatedpotassiumlevelson cardiac conduction since they interfere with repolarization of the cellular membrane. ECG changes appear progressively
Table 5.8: Causes of hyperkalemia Decreased losses
Renal failure
Renal tubular disorders: Pseudohypoaldosteronism, urinary tract obstruction
Drugs: ACE inhibitors, angiotensin receptor blockers, potassium sparing diuretics, NSAIDS, heparin Mineralocorticoid deficiency: Addison disease, 21-hydroxylase deficiency, 3P-hydroxysteroid dehydrogenase deficiency
Increased intake
Intravenous or oral potassium intake; packed red cells transfusion
Extracellular shift
Acidosis, low insulinstate, medications (P-adrenergic blockers, digitalis, succinylcholine, fluoride), hyperkalemic periodic paralysis, malignant hyperthermia
Cellular breakdown
Tumor lysissyndrome, rhabdomyolysis, crush injury, massive hemolysis
with rising serum potassium and include tall, peaked T waves (5.5 to 6.5 mEq/1), prolonged PR interval, flat P waves, wide QRS complex (6.5 to 8.0 mEq/1), absent P waves, bundle branch blocks and eventually sine waves (>8.0 mEq/1).
Treatment
Hyperkalernia is a medical emergency, requiring prompt discontinuation of potassium-containing fluids and administration of medications that ensure stability of myo cardial membrane, intracellular shift of potassium and enhance its elimination (Box 5.4). Continuous ECG moni toring should be performed. Treatment should be individualized based upon the presentation, potassium level, and ECG changes. If the hyperkalemia is severe (potassium >7.0 mEq/1) or thepatient is symptomaticwith ECG changes, therapy should be initiated promptly with intravenous calcium gluconate, followed by sodium bicarbonate, insulin-glucose infusion and/or nebulized2-agonists. Hemodialysis may be needed in the more refractory patients. Milder elevations (5.5-6.5 mEq/1) are managed with elimination of potassium intake, dis continuation of potassium sparing drugs and treatment of the underlying etiology. Children with primary or secondary hypoaldosteronism require stress-dose steroid supplements and mineralocorticoids.
Sox 5.4: Treatment of hyperkalemia
•Prompt discontinuation of potassium-containing fluids and medications that lead to hyperkalemia
•Stabilize the myocardial cell membrane to prevent lethal cardiac arrhythmia. Use intravenous (IV) 10% calcium gluconate (or calcium chloride), at 0.5 ml/kg over 5-10 minutes under cardiac monitoring. Discontinue if bradycardia develops
•Enhance cellular uptake of potassium
Regular insulin and glucose IV: (0.3 D regular insulin/g glucose over 2 hr)
Sodium bicarbonate IV: 1-2 mEq/kg body weight over 20-30 minutes
Beta-adrenergic agonists, such as salbutamol and terbutaline nebulized or IV
•Ensure total body potassium elimination
Sodium polystyrene sulfonate (Kayexalate) oral/per rectal: 1 g/kg (max. 15 g/dose) oral or as rectal enema in 20-30% sorbitol
Loop or thiazide diuretics (only if renal function is maintained)
•Hemodialysis is necessary to treat severe symptomatic
hyperkalemia that is resistant to drug therapy, particularly in patients with impaired renal functions. Continuous veno-venous hemofiltrationwithdialysis(CVVHDF)have also been used to remove potassium
•Children with primary or secondary hypoaldosteronism require maintenance steroids and mineralocorticoid supplements
CALCIUM
Physiology
Ninety-eight percent of body calcium is found in the skeleton which is in equilibrium with the extracellular concentration of calcium. Approximately 1 to 2% of body calcium exists in the ECF for physiological functions like blood coagulation, cellular communication, exocytosis, endocytosis, muscle contraction and neuromuscular transmission. Calcium affects the intracellular processes, throughitscalcium-bindingregulatory protein,calmodulin.
Most of the filtered calcium is reabsorbed in the proximal tubule (70%), ascending loopof Henle(20%)and the distal tubule and collecting duct (5-10%). Factors that promote calcium reabsorption include parathormone (PTH), calcitonin, vitamin D, thiazide diuretics and volume depletion. Volume expansion, increased sodium intake and diuretics such as mannitol and frusemide promote calcium excretion.
The intestine serves as a longterm homeostatic mechanism for calcium. Although the major source of calcium is dietary, less than 15% of dietary calcium is absorbed, primarily in the ileum and jejunum by means of active transport and facilitated diffusion. Calcium is controlledprimarily by majorregulatoryhormones,PTH, calcitonin andvitaminD. Additionally thyroidhormones,
Fluid and Electrolyte Disturbances -
growth hormone, adrenal and gonadal steroids also have minor influences on calcium metabolism.
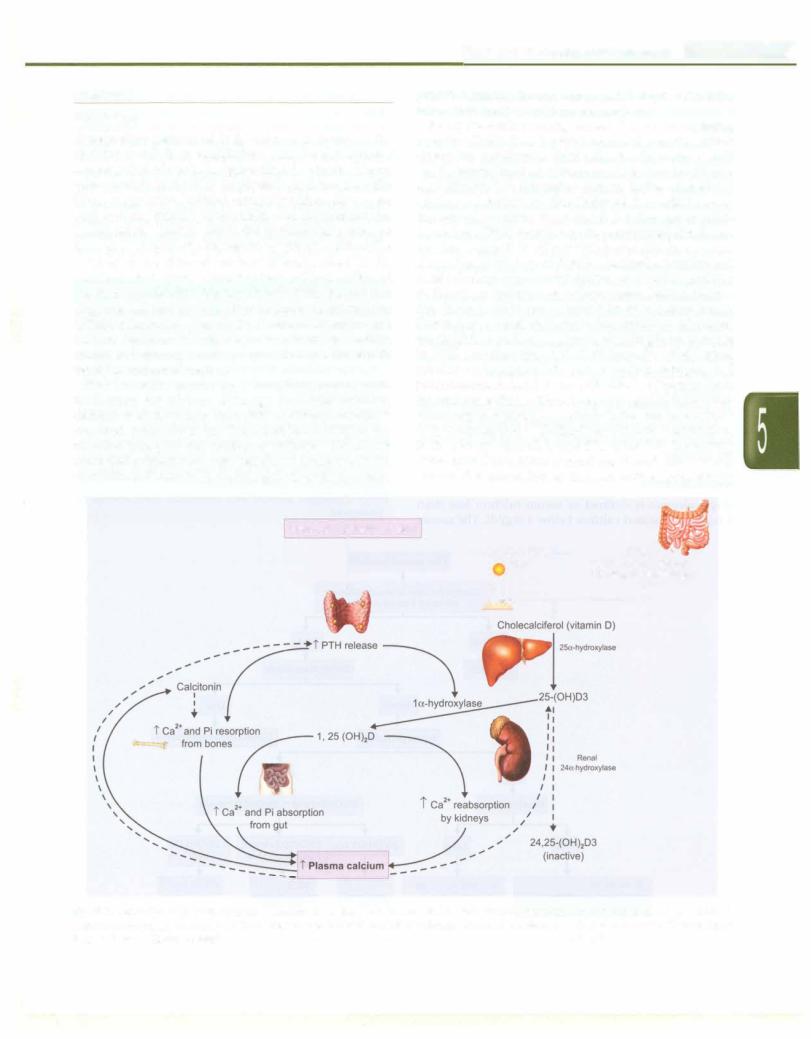
Role ofthe calcium-sensing receptor. The calcium-sensing receptor (CaSR) is a G protein-coupled receptor, which allows the parathyroid chief cells, the thyroidal C cells and the ascending limb of the loop of Henle (renal tubular epithelial cells) to respond to changes in the extracellular calcium concentration. The ability of the CaSR to sense the serum calcium is essential for the appropriate regulation ofPTH secretion by the parathyroid glands and for the regulation of passive paracellular calcium absorption in the loop of Henle. Calcitonin secretion and renal tubular calcium reabsorption are directly regulated by theactionofcalciumion onitsreceptor.Ionizedcalcium acts through calcitonin, to inhibit its release from bones. Decreaseinextracellularcalciumconcentration, stimulates the CaSR in parathyroid glands, resulting in an increase in PTH secretion (Fig. 5.7). PTH increases distal renal tubular reabsorption of calcium within minutes and stimulates osteoclast activity, withreleaseofcalciumfrom the skeletonwithin1-2 hr. More prolongedPTH elevation stimulates la-hydroxylaseactivityin theproximaltubular cells, which leads to 1, 25-dihydroxy-vitaminD production. In the kidney, vitamin D and PTH stimulate the activity of the epithelial calcium channel and the calcium-binding protein (i.e. calbindin) to increase active transcellular
Decreased p asma calcium |
7-dehydrocholesterol in |
Dietary |
||
|
||||
l |
|
skin |
Vit D2 (ergocalciferol) |
|
{ |
1 I |
Vit D3 (choralciferol) |
||
|
{{ff·,fr |
- i------ |
||
Fig. 5.7: Regulation of plasma calcium. Reduction in ionized calcium results in parathormone secretion, which through direct and indirect actions on the bones, intestine and the kidneys results in positive calcium balance. Calcitonin results in accretion of bone mass. Discontinuous lines indicate inhibitory control

|
l |
_____________________________ |
E_s_s_ e_tn__ia_P_ed_iat _ris__c |
||
__ |
|
|
calcium absorption in the distal convoluted tubule. These mechanisms help to maintain normal levels of serum calcium.
Plasma calcium exists in 3 different forms: 50% as biologically active ionized form, 45% bound to plasma proteins (mainlyalbumin)and5%complexedtophosphate and citrate. In the absence of alkalosis or acidosis, the proportion of albumin-bound calcium remains relatively constant. Metabolic acidosis leads to increased ionized calcium from reduced protein binding and alkalosis has the opposite effect. Plasma calcium is tightly regulated despite its large movements across the gut, bone, kidney and cells in the normal range of 9-11 mg/dl.
Because calcium bindsto albumin and onlythe unbound (free or ionized) calcium is biologically active, the serum level must be adjusted for abnormal albumin levels. For every1 g/dldropinserumalbuminbelow4 g/dl,measured serum calcium decreases by 0.8 mg/dl. Corrected calcium can be calculated using the following formula:
Corrected Ca =
[4 - plasma albumin in g/dl] x 0.8 + measured serum calcium
Alternatively, serum free (ionized) calcium levels can be directly measured, negating the need for correction for albumin.
Hypocalcema i
Hypocalcemia is defined as serum calcium less than 8 mg/dl or ionized calcium below 4 mg/dl. The causes
and algorithm for investigating the etiology are shown in Table 5.9 and Fig. 5.8.Hypocalcemia manifests as central nervous system irritabilityandpoormuscular contractility. Newborns present with nonspecific symptoms such as lethargy, poor feeding, jitteriness, vomiting, abdominal distension and seizures. Children may develop seizures, twitching, cramps and rarely laryngospasm (Box 5.5). Tetany and signs of nerve irritability may manifest as musculartwitching, carpopedalspasmandstridor.Latent tetany can be diagnosed clinically by clinical maneuvers such as Chvostek sign (twitching of the orbicularis oculi
Table 5.9: Causes of hypocalcemia
Neonatal: Early (within 48-72 hr after birth) or late (3-7 days after birth) neonatal hypocalcemia; prematurity; infant of diabetic mother; neonates fed high phosphate milk Parathyroid: Aplasia or hypoplasia of parathyroid glands, DiGeorge syndrome, idiopathic; pseudohypoparathyroidism; autoimmune parathyroiditis; activating mutations of calcium sensing receptors
Vitamin D: Deficiency; resistance to vitamin D action; acquired or inherited disorders of vitamin D metabolism
Others: Hypomagnesernia; hyperphosphatemia (excess intake, renal failure); malabsorption syndromes; idiopathic hypercalciuria; renal tubular acidosis; metabolic alkalosis; hypoproteinemia; acute pancreatitis
Drugs: Prolonged therapy with frusernide, corticosteroid or phenytoin
|
|
|
|
|
|
|
• |
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
Low serum calcium |
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
Correct for level of serum albumin; |
|
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
• |
|
measure ionized calcium |
J_ |
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
Normal |
|
|
|
|
|
|
Low |
|
|
|
|
|
|
|
|||
|
|
|
|
No action] |
|
|
|
|
|
|
+ |
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
Serum magnesium |
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
Normal |
|
|
|
|
|
|
LowJ |
|||||||
|
|
|
+ |
|
|
Serum phosph |
+ Correct hypomagnesemiaj |
||||||||||||||
|
|
|
|
|
|
|
= |
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
• |
|
|
|
|
|
|
|
|
.r |
|
|
|
|
|
|
|
||
|
|
|
High |
|
|
|
|
|
|
|
|
Low |
|
|
|
|
l |
||||
|
|
|
Parathormone |
|
|
|
|
|
l?5(0H)D3and 1,25(0H) |
2 |
|
|
|
||||||||
|
+ |
|
|
|
|
|
|
D3 levels |
|||||||||||||
|
I |
+ |
|
|
+L. --F |
|
|
|
|
• |
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
Hi |
|
|
Low |
|
OH)D |
3 low ll |
|
25(0H)D3 normal |
I |
1,25(0HhD3 |
||||||||||
|
|
|
|
|
1,25(0HhD3 low |
|
|
|
high |
||||||||||||
|
|
|
r |
|
|
|
|||||||||||||||
Renal failure |
! |
|
|
|
|
+ |
|
|
+ |
|
|
+ |
|
|
|||||||
|
|
|
|
|
|
|
|
Vitamin D |
VDDR type I |
|
|
VDDR type II |
|||||||||
Pseudohypoparathyroidism |
|
|
|
|
deficiency |
Renal failure |
|
|
|
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
Tumor lysis |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Fig. 5.8: Algorithm for evaluation of hypocalcemia. VDDR vitamin D dependent rickets

Box. 5.5: Clinical features of hypocalcemia
•Carpopedal and muscle spasms
•Tetany
•Laryngospasm
•Paresthesias
•Seizures
•Irritability, depression, psychosis
•Intracranial hypertension
•Prolonged QTc interval
and mouth elicited by tapping the facial nerve anterior to the external auditory meatus) and the Trousseau sign (carpopedal spasm elicited by inflating a blood pressure cuff on the arm to a pressure above the systolic pressure for 3 min). ECG shows prolonged corrected QT interval (QTc) to more than 0.45 seconds. Cardiac function may be impaired because of poor muscle contractility. Prolonged hypocalcemia can present with features of rickets.
Management
Tetany, laryngospasm and seizures must be treated immediately with 2 ml/kg of 10% calcium gluconate, administeredIV slowlyunder cardiac monitoring.Calcium gluconate 10% (100 mg/ml) IV solution contains 9.8 mg/ ml (0.45mEq/ml) elementalcalcium; calciumchloride 10% (100 mg/ml)contains 27 mg/ml (1.4 mEq/ml). Initially IV calcium boluses are given every 6 hr. Thereafter, oral calcium supplementation is provided at 40-80 mg/kg/ day. Oral calcium therapy is used in asymptomatic pati ents and as followup to intravenous (IV) calciumtherapy. Intravenous infusion with calcium-containing solutions can cause severe tissue necrosis; therefore integrity of the IV site shouldbe ascertained before administering calcium through a peripheral vein. Rapid infusion of calcium containing solutions through arterial lines can cause arterial spasm and if administered via an umbilical artery catheter, intestinal necrosis. Magnesium administration is necessary to correct any hypomagnesemia because hypocalcemia does not respond until the low magnesium level is corrected. In patients with concurrent acidemia, hypocalcemia should be corrected first. Acidemia increa ses the ionized calcium levels by displacing calcium from albumin. If acidemia is corrected first, ionized calcium levels decrease.
Calciumcarbonateis an oralsupplementproviding40% elemental calcium. Therapy with cholecalciferol is used in patients withvitamin Ddeficiency.Calcitriol, an active metabolic form of vitamin D (i.e. 1,25-dihydroxychole calciferol) is administered in liver or renal disease.
Hypercalcemia
Hypercalcemia is defined as a serum calcium level greater than 11 mg/dl. Because calcium metabolism normally is
Fluid and Electrolyte Disturbances -
tightly controlled by the body, even mild persistent elevations should be investigated. Etiologies of hyper calcemia vary by age and other factors (Table 5.10). Hypercalcemiaisoftenasymptomatic,althoughitcancause symptoms at levels as low as 12 mg/dl and consistently at values above 15 mg/dl. Such high values are however, rarely encountered and present as stupor and coma. Neonates may be asymptomatic or may have vomiting, hypotonia,hypertensionorseizures.Clinicalfeatures inolder children are summarized in Box 5.6 and include irritability,
Box 5.6: Clinical features of hypercalcemia
Lethargy, confusion, |
Bradycardia, systemic |
depression, coma |
hypertension, headache |
Hyporeflexia |
Nephrocalcinosis, |
Muscle weakness |
nephrolithiasis |
Constipation |
Polyuria |
|
Reduced QTc interval |
malaise, headache, confusion, unsteady gait and proximal muscle weakness. Abdominal pain with paralytic ileus, nausea and vomiting and constipation are often observed. Ectopic calcification can lead to symptoms of pancreatitis, with epigastric pain and vomiting. Ectopic calcification can manifest as conjunctivitis or band keratopathy. Renal manifestationsdueto renalstonesandnephrocalcinosiscan progress to renalfailure,and polyuria andpolydipsia occur due to nephrogenic diabetes insipidus.
Treatment
The initial treatment of hypercalcemiainvolves hydration to improve urinary calcium excretion. Rapid lowering of serum calcium can be expected with isotonic sodium
Table 5.10: Causes of hypercalcemia
Neonates
Neonatal primary hyperparathyroidism, secondary hyper- parathyroidism
Familial hypocalciuric hypercalcemia Excessive supplementation of calcium
William syndrome, hypophosphatasia, idiopathic infantile hypercalcernia
Older children
Hyperparathyroidism (parathyroid adenoma, autosomal dominant hereditary hyperparathyroidism, multiple endo crine neoplasia type 1)
Malignancies: Non-Hodgkin or Hodgkin lymphoma, Ewing sarcoma, neuroblastoma, Langerhans cell histiocytosis, rhabdomyosarcoma
Granulomatous disease: Sarcoidosis, tuberculosis, Wegener disease, berylliosis
Others: Vitamin Dor A intoxication; thiazide diuretics; milk alkalisyndrome; dietary phosphatedeficiency; subcutaneous fat necrosis; thyrotoxicosis; prolonged immobilization

__E_s_se_n_t_i_a_l _P_e _d_ iat _r_ic_ s-------------------------- |
-------- |
_ |
|
chloride solution, because increasing sodium excretion increases calcium excretion. Addition of a loop diuretic inhibits tubular reabsorption of calcium but attention should be paid to other electrolytes (e.g. magnesium, potassium)duringsalinediuresis. Bisphosphonates serve toblockboneresorptionand decreaseserumcalciumwithin a couple of days but have not been used extensively in children. Pamidronate and etidronate have been used in the treatment of hypercalcemia due to malignancy, immobilization and hyperparathyroidism but may cause mineralization defects.
Peritoneal dialysis or hemodialysis can be used in extreme situations, particularly in patients with renal failure.Calcimimetics (cinacalcet hydrochloride)changethe configuration of the CaSR in a manner that makes it more sensitive to serum calcium. Its safety and efficacy in pediatric population has yet to be substantiated. Surgical intervention may be needed in patients with hyper parathyroidism, particularlywith recurrent renal stones or persistent serum calcium levels higher than 12.5 mg/dl.
Subtotalparathyroidectomycanbe performed, or complete parathyroidectomy can be chosen with reimplantation of a small amount of tissue in the forearm.
MAGNESIUM
Physiology
Magnesiumis thethird-most abundantintracellularcation predominantly located in muscle and liver cells. Most intracellular magnesium is bound to proteins; only approximately 25% is exchangeable.
Magnesiumplays a fundamental role in manyfunctions ofthe cell,including energytransfer andstorageand nerve conduction. Magnesium also plays important role in protein, carbohydrate, and fat metabolism, maintenance of normal cell membrane function and regulation of PTH secretion.
Dietary sources include green leafy vegetables, cereals, nuts and meats. Absorption of magnesium takes place primarily in the small intestine and is inversely related to the amount of magnesium, calcium, phosphate and fat. PTH and glucocorticoids increase magnesium absorption. Absorption is diminished in presence of substances that complex with magnesium (free fatty acids, fiber, phytate, phosphate, oxalate); increased intestinal motility and calcium also decrease magnesium absorption. Vitamin D and PTH enhance absorption. Renal excretion is the principal regulator of magnesium balance. Reabsorption occurs chiefly in the thick ascending loop of Henle (70%) and to a smaller extent in the proximal (15%) and distal (5-10%) tubules. Fractional excretion of magnesium exceeding 4% indicates renal magnesium wasting.
Hypomognesemio
Hypomagnesemia develops from decreased intake or more commonly increased losses which could be
gastrointestinal (diarrhea, vomiting, nasogastric suction) or renal (chronic use of thiazide diuretics, recovery phase of acute tubular necrosis, Gitelman syndrome, familial hypomagnesemia-hypercalciuria-nephrocalcinosis).
Symptomatic magnesium depletion (occursatlevelsbelow
1.2 mg/dl) is often associated with multiple biochemical abnormalities,including hypokalemia, hypocalcemia and metabolic acidosis. As a result, hypomagnesemia is sometimes difficult to attribute solely to specific clinical manifestations. Hypomagnesemia often leads to hypo calcemia, possibly by inhibition of PTH activity. Neuromuscular manifestations of hypomagnesemia include muscle weakness, tremors, seizures, paresthesias, tetany, positive Chvostek sign, Trousseau signs and nystagmus. Cardiovascular manifestations include nonspecific T-wave changes, U-waves and prolonged QT interval and arrhythmias.
Treatment
Therapy can be oral for patients with mild symptoms or intravenous for patients with severe symptoms or those unable to tolerate oral administration. Severe hypoma gnesemia is treated with slow intravenous infusion of magnesium sulfate (50% solution) at adoseof 25-50 mg/kg (2.5-5.0 mg/kg of elemental magnesium). The dose is repeated every6 hr, for a total of 2-3 doses. Doses needtobe reduced in children with renal insufficiency. Oral replace ment should be given in the asymptomatic patient, or those requiring longterm replacement, preferably with a sustained-release preparation to avoid diarrhea. Oral magnesiumpreparationprovide5-7mEqofmagnesiumper tablet. Two to four tablets may be sufficient for mild, asymptomatic disease while severe cases require up to six to eight tablets, to be taken daily in divided doses. Patients with renal magnesium wasting may benefit from diuretics with magnesium-sparingproperties,such as spironolactone and amiloride.
Hypermognesemio
Serum magnesium >2.5 mg/dl is uncommon in children and may be seen in the setting of renal insufficiency, prolonged use of magnesium containing antacids or in neonates born to mothers given magnesium sulfate as a treatment for eclampsia. Symptoms of hypermagnesemia are nonspecific at lower levels: nausea, vomiting,flushing, lethargy, weakness and dizziness. At higher levels, deep tendonreflexesaredepressed which may progress to coma and respiratory depression. Effects on the heart may result in prolongation of intervals on ECG or manifest as arrhythmias, complete heart block and asystole.
Treatment
In patients with mildly increased levels, the source of magnesium maysimplyberemoved. Intravenous calcium directlyantagonizes the cardiac and neuromuscular effects of excess extracellular magnesium. Dialysis may be used

for patients with severe hypermagnesemia and renal impairment, or those with serious cardiovascular or neuromuscular symptoms.
ACID-BASE DISORDERS
Regulation of Acid-Base Equilibrium
The body is sensitive to changes in blood pH level, as disturbances in acid-base homeostasis can result in denaturation ofproteinsandinactivation ofenzymes that may be potentially fatal. Strong mechanisms exist to regulate acid-base balance and maintain arterial pH (7.35 to7.45),pC02 (35 to 45 mmHg)andHC03-(20to28mEq/1) within a narrow range.
Acidemia is defined as H+concentration exceeding 45 nmol/1 (pH <7.35); values below 35 nmol/1 (pH>7.45) define alkalemia. The disorders that cause acidemia or alkalemia are termed asacidosis and alkalosis respectively. Metabolic activity results in production of two types of acids, carbonic acid (a volatile acid, derived from carbon dioxide) and nonvolatile acids (including sulfuric acid, organic acids, uric acid and inorganic phosphates). Accumulation ofH+ ions of nonvolatileacidsduetoexcess production or inadequate buffering, failure to excrete H+
Fluid and Electrolyte Disturbances -
or loss of bicarbonate results in metabolic acidosis. If the reverse occurs, it results in metabolic alkalosis. The principle mechanism for carbon dioxide handling is by the lungs. Hyperventilation results its CO2 washout and drop in arterial pC02 (respiratory alkalosis), hypo ventilation has the opposite effect (respiratory acidosis). Whenonly oneprimaryacid-baseabnormality occursand its compensatory mechanisms are activated, the disorder is classified as simple acid-base disorder. A simple algorithmfordefiningsimpleacid-basedisordersisshown in Fig. 5.9. When a combination of disturbances occurs, the disorder is classified as a mixed acid-base disorder. Thelatteraresuspectedwhenthecompensationinagiven patient differs from the predicted values in Table 5.11.
In order to maintain body homeostasis, changes in pH are resisted by a complex system of intracellular and extracellular buffers. The first line of defense, are the chemical buffers. In metabolic disorders the extracellular buffersrapidlytitratetheadditionofstrongacidsorbases. Intracellular buffers chiefly accomplish the buffering of respiratory disorders. Secondary respiratory compen sations to metabolic acid-base disorders occur within minutes and is completed by 12 to 24 hr. In contrast secondary metabolic compensation of respiratory
|
|
|
|
|
|
|
|
--, |
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
Blood pH |
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Acidemia |
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
(l pH) |
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
t |
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Lr |
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
IRespiratory acidosis |
|
Metabolic acido |
Respiratory alkalosis Metaboli |
|||||||||||||||
|
|
|
|
|
Metabolic acidosis |
|
|
|
|
|
|
|
||||||
|
Respiratory acidosis |
|
|
|
|
Metabolic alkalosis |
||||||||||||
|
with compensatory |
|
with compensatory |
|
|
|
with compensatory |
|||||||||||
|
metabolic alkalosis |
respiratory alkalosis |
|
|
respiratory acidosis |
|||||||||||||
|
|
|
Fig. 5.9; Algorithm for simple acid-base disorders |
|
|
|||||||||||||
|
|
Table 5.11: Compensation for primary acid-base disorders |
||||||||||||||||
Disorder |
Primary event |
|
|
Compensation |
Expected Compensation |
|||||||||||||
Metabolic acidosis |
l [HC03-J |
|
|
l pC02 |
pC02 l by 1-1.5 mm Hg for 1 mEq/l l[HC03] |
|||||||||||||
Metabolic alkalosis |
t [HC03-J |
|
|
t pC02 |
pC02 t by 0.5-1 mm Hg for 1 mEq/1 t [HC03] |
|||||||||||||
Respiratory acidosis |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Acute (<24 hr) |
|
tpC02 |
|
|
t[HC03-J |
[HC03] t by 1 mEq/1 for 10 mm Hg t pC02 |
||||||||||||
Chronic (3-5 days) |
|
tpC02 |
|
|
ft [HC03-J |
[HC03-J t by 4 mEq/1 for 10 mm Hg t pC02 |
||||||||||||
Respiratory alkalosis |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Acute (<24 hr) |
|
lpC02 |
|
|
l[HC03-J |
[HC03-J l by 1-3 mEq/1 for 10 mm Hg l pC02 |
||||||||||||
Chronic (3-5 days) |
|
lpC02 |
|
|
U[HC03-J |
[HC03-J l by 2-5 mEq/1 for 10 mm Hg l pC02 |
||||||||||||
___E_s_s_e_n_ t_ia_l_P_e_d_i_a _tr-ics_________________________________
disorders begins more slowly and takes 2 to 5 days for completion.The compensatory mechanisms do not return the pH to normal until the underlying disease process has been appropriately treated. Several buffering agents reversibly bind hydrogen ionsand impede any change in body pH. Extracellular buffers include bicarbonate and ammonia, whereas proteins and phosphate act as intracellular buffers.
Bicarbonate-carbonic acid buffer in the extracellular fluid, is the key buffer as carbon dioxide (CO2) can be shifted through carbonic acid (H2C03) to hydrogen ions and bicarbonate (HC03-):
HP + CO2 H H2C03 HH++ HC03.
Acid-base imbalances that overcome the buffer system can be compensated in the short-term by altering the rate of ventilation, which alters the pC02• While this is a rela tivelyweakbuffer,itaccountsfor55%ofthebufferingcapa citybecauseofitssheerabundance. WhenH+ concentration increases the above reaction shifts to the left, more CO2 is generated and exhaled from the lungs, moderating the change in pH. Hemoglobin is a powerful intracellular buffer because its negatively charged histidine moieties accept H+, normalizing the pH. Other proteins also have negative charges that can accept H+.
Renal Regulation of Acid-base Balance
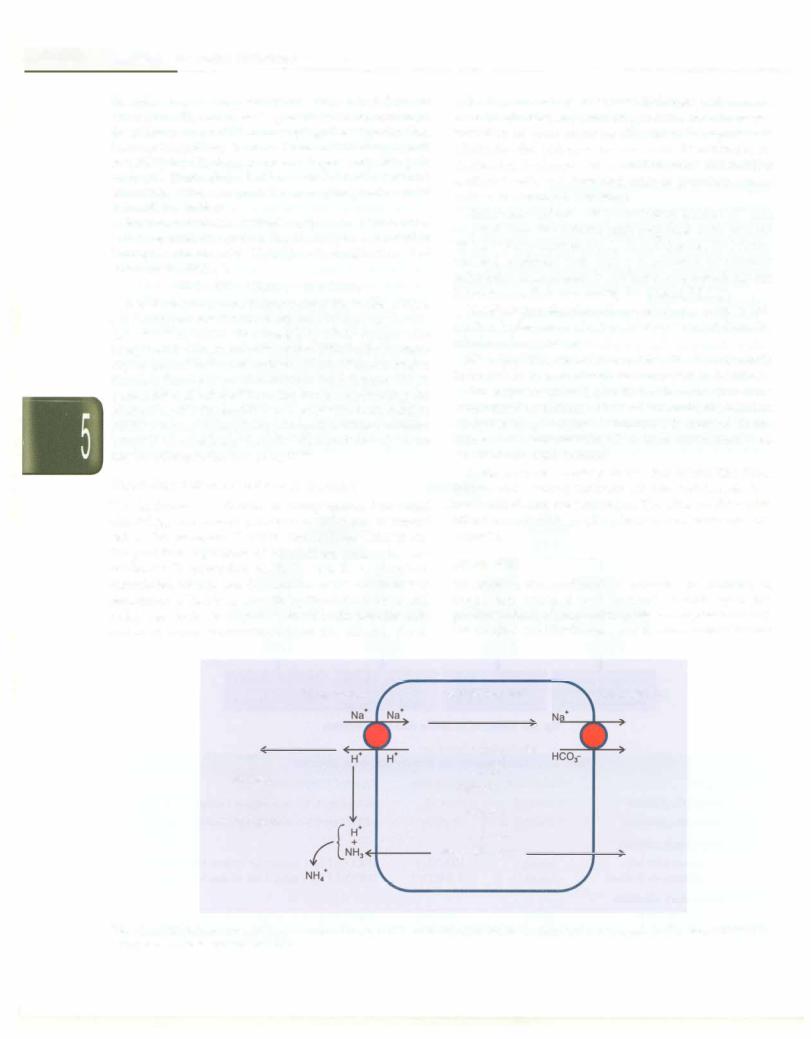
The kidneys are slower to compensate, but renal physiology has several powerful mechanisms to control pH by the excretion of excess acid or base. Kidneys are the principal regulators of bicarbonate mainly by two methods: (i) resorption of HC03- mostly in proximal convoluted tubules and (ii) excretion of H+ and therefore generation of HC03, primarily by the distal tubules and collecting ducts. In responses to acidosis, tubular cells reabsorb more bicarbonate from the tubular fluid,
collecting duct cells secrete more hydrogen and generate morebicarbonate, andammoniagenesis leads to increased formation of renal ammonia (Fig. 5.10). In responses to alkalosis, the kidneys excrete more bicarbonate by decreasing hydrogen ion secretion from the tubular epithelial cells, and lowering rates of glutarnine meta bolism and ammonia excretion.
Bicarbonate-carbonic acid in the kidney tubules: H+ ions secreted from the tubular cells combines with luminal
HC0 - to form water and CO2. The CO2 enters the tubular
3
cell and combines with water, in presence of carbonic anhydrase, to regenerate HC03thatis reabsorbed into the bloodstream, thus conserving the filtered HC03.
Monohydrogen phosphate-dihydrogenphosphate buffer: This luminal buffer system buffers H+ ions secreted from the tubular cells, as follows:
H+ + Na2HP04 NaH2P04 + Na+. The weakly acidic sodium dehydrogen phosphate is excreted in the urine.
Ammonia-ammonium buffer: In tubular cells glutarnine is converted to glutamic acid and ammonia, the reaction catalyzed by glutaminase. Ammonia is secreted in the lumen and combines with H+ to form ammonium ions, which are excreted in urine.
Sodium-hydrogen exchange in the distal tubule: The distal tubular cells actively reabsorb Na+; to maintain electro neutrality H+ ions are exchanged. Thelatter combine with either monohydrogen phosphate or ammonia and are excreted.
Anion Gap
To achieve electrochemical balance, the number of negatively charged ions (anions) should equal the positivelycharged ions (cations). Measuredplasmaanions are chloride and bicarbonate, and the unmeasured anions
Tubular lumen
H+
+
Na2HP04
1{ )
NaHp04+ Na+
Renal tubule cell |
Interstitial fluid |
H+ + HC03--+
1t-n}
H20+C02
Glutamine
l (i:,\...,...
NH3 + Glutamate
Fig. 5.10: Renal regulation of acid-base disorders. 1 = bicarbonate-carbonic acid buffer; 2 = monohydrogen phosphate-dihydrogen phosphate buffer; 3 = ammonia-ammonium buffer