

Ghai Essential Pediatrics8th
.pdfimplies a deranged response of renal la-hydroxylase to a low phosphate signal.
Clinical features Limb deformities such as coxa vara, genu valgum, genu varum and short stature may occur. Abnormalities of maxillofacial region and premature fusion of cranial sutures maylead to deformities of skull. Dental abnormalities are commonly seen including pulp deformities with intraglobular dentine, and frequent dentalabscesses. Symptoms of hypocalcemia (tetany and muscle weakness) are absent. The mother of affected patient(s) may have bowing of legs and short stature or fasting hypophosphatemia.
Evaluation Changes of active rickets in spine and pelvis arerarelyseeneveninadvancedstages.Thelevelofserum calcium is normal or slightly low (9-9.5 mg/dl), that of phosphate decreased (1.5-3 mg/dl). Serum alkaline phosphatase levelis raised.PTHlevelsare normal. Blood levelsof l,25(0H)iD3areinappropriately lowforthelevel of serum phosphate. Urinary phosphate excretion is increased with decreased tubular reabsorption of phos phate.
Management Oral phosphate and vitamin D supple ments are administered. Phosphates are provided in a dosageof30-50mg/kg(total1-3gelementalphosphorus) divided into 5 to 6 equal parts and can be given in the formofJouliesolutionorasneutralphosphateeffervescent tablets.Joulie solution contains 30.4mg of phosphate/ml. Diarrhea is a frequent problem with higher doses.
Vitamin D supplementation is necessaryfor healing of rickets.Treatment isstartedwith alpha-calcidiolatadose of 25-50 ng/kg/day (maximum 2 µg/day) until there is biochemicalandradiologicalevidenceofhealingofrickets. Periodic monitoring of serum and urine levels of calcium and phosphate is essential. A level of serum phosphate greater than 3.0 to 3.2 mg/dl is desirable.
Vitamin D Dependent Rickets (VDDR)
Theserareautosomalrecessivelyinheritedrickets areseen in infants between 3 and 6 months of age, who have been receiving the usual amounts of vitamin D. Two forms are seen.
VDDR type I This condition is characterized by a deficiency of the enzyme, 25-hydroxyvitamin D la hydroxylase. Reduced blood levels of calcium, normal to low phosphate and elevated alkaline phosphatase are characteristic. Blood levels of 25(0H)D3 are normal but those of 1, 25(0H)iD3 are markedly decreased despite hypocalcemia.
Theclinicalfeatures are similar to vitamin Ddeficiency rickets and include hypotonia, growth failure, motor retardation (poor head control, delayed standing and walking), convulsions due to hypocalcemia, anemia and occasionally respiratory difficulty. Physical examination
Micronutrients in Health and Disease -
shows thickening of wrists and ankles, frontal bossing, widely open anterior fontanelle, rickety rosary, bony deformities and positive Trousseau and Chvostek signs. Dentition is delayed and development of tooth enamel impaired.
The treatment of VDDR type I is with physiological doses of alpha-calcidiol or calcitriol (1-2 µg daily). Most subjectsrequireconcomitant treatmentwithcalciumwith or without phosphate supplements. With appropriate therapy the serum calcium levels rise and radiological healing occurs within 6 to 8 weeks.
VDDR type II The features are similar to VDDR type I. There is end organ resistance to 1,25(0H)iD3. This leads to virtual abolition of actions of l,25(0H)iD3, despite its markedly raised levels in circulation (secondary to hypocalcemia and low 24-hydroxylase activity).
Earlyonsetofrickets, ahighprevalenceofalopeciaand ectodermal defects (oligodontia, milia and epidermal cysts)are characteristic.Hypocalcemia, secondaryhyper parathyroidism, elevated circulating levels of l, 25(0H)iD3 and an absence or decreased response to vitamin D analogs are seen. The response to treatment in patients with VDDR type II is not satisfactory. An occasional patient may get clinical and biochemical improvementandradiologicalhealingfollowinglongterm administration of large amounts of intravenous or oral calcium.
Other Causes of Rickets
Renal tubular acidosis Proximal or distal renal tubular acidosis (RTA) are important causes of refractory rickets in children. The conditions are characterized by hyper chloremic metabolic acidosis with normal blood levels of urea and creatinine. Patients with proximal RTA may show low blood levels of phosphate, aminoaciduria and low molecular weightproteinuria. The useof bicarbonate andphosphatesupplementation(inproximalRTA)results in healing of rickets.
Chronic kidney disease Refractory rickets may occa sionallybethepresentingmanifestationof chronickidney disease (GFR below 30-35 ml/rnin/1.73 m2), particularly in patients with tubulointerstitialdisease. The features of mineral bone disease depend on patient ageandduration of disease. Elevated blood levels of creatinine, phosphate and parathormone are characteristic. Therapy consists of restricting phosphateintake and providing supplements of calcium and active vitamin D analogs.
Oncogenous rickets Benign mesenchyrnal tumors may secrete fibroblast growth factor that results in phos phaturia, hypophosphatemia, rickets and muscle weak ness. The tumor may be small and difficult to detect but its removal reverses the biochemical abnormalities and heals the rickets.

-......E_s_s_e_n_t _ia_i_P_e_d_i_at_r_ics_________________________________
Metaphyseal dysplasia Several types of disorders are described.Short stature with bowing of legs and waddling gait are prominent. There are no biochemical abnor malities except for occasional hypercalcernia in Jansen metaphyseal chondrodysplasia. Radiological changes resemble rickets.There is no treatment for these disorders. Fluorosis Endemic fluorosis might present with bony deformities and radiological features of rickets in school going children. Pain in limbs and spine, mottling of teeth and familyhistoryofasimilarillnessareimportantfeatures. Osteosclerosis and calcification of ligaments may be found in older children and adults; levels of alkaline phosphatase and parathormone are raised. Levels of fluoride are increased in the water consumed, urine and blood.
VitaminE
Vitamin E (tocopherol) functions as a membrane bound antioxidant. Only 20-40% of ingested tocopherol and/or its esters are absorbed. The absorption is enhanced by simultaneous digestion and absorption of dietary lipids. Medium chain triglycerides,and bile and pancreaticjuices enhance absorption of tocopherol, which is incorporated intochylomicronsanddelivered to theliver.Fromthe liver it is secreted with VLDL and LDL and delivered to peripheral tissues. Red blood cells, which contain about
20% of vitamin E in plasma, also participate in transport.
Nutritional requirements Vitamin E requirement of normal infants is approximately 0.4 µg/kg body weight/ day. For premature infants, 15 to 20 mg/day is required. The RDA for infants increases from 3 to 6 mg tocopherol from birth to 2 yr of age. One mg of tocopherol provides
1.5 IU activity of vitamin E.
Sources The common sources of vitamin E are vegetable oils (com, cottonseed, safflower) and margarine. Other sourcesinclude leafy vegetables and nuts; breast milk and colostrum are rich sources.
Vitamin E Deficiency
Infants particularly if prematures are born in a state of relative tocopherol deficiency. This is attributed to limited placental transfer of vitamin E, relative dietarydeficiency, intestinal malabsorption and rapid growth. The risk of vitamin E deficiency is increased in infants fed onformulae high in polyunsaturated fats and low tocopherol content. As the digestive system matures, tocopherol absorption improves and itsbloodlevels rise.Acommonpresentation of the deficiency is in preterm babies in hemolytic anemia. The levels of hemoglobin range between 7 and 9 g/dl. Reticulocytosis and hyperbilirubinernia are accompanied by low levels of vitamin E. Administration of iron exacerbates hemolysis, unless vitamin E is also adminis tered. Parenteraltherapywith vitamin E improves anemia and corrects hemolysis.
A commoncauseofvitaminE deficiencyin olderchildren and adolescentsis fat malabsorption.Abetalipoproteinemia,
caused by the genetic absence of apolipoprotein B,causes fat malabsorption and steatorrhea, with progressive neuropathy and retinopathy in the first two decades of life. Plasmalevelsare undetectable and high dose vitamin E improves neurologicalsymptoms. Other manifestations include the neurologicsyndrome of spinocerebellarataxia with loss of deep tendon reflexes, truncal and limb ataxia, loss of vibration and position sense, ophthalmoplegia, muscle weakness, ptosis and dysarthria. A pigmented retinopathy may also occur.
Isolated form of vitamin E deficiency without fat malabsorption has been reported. Most malabsorption syndromes respond to large doses of oral vitamin E (100-200mg/kg/day) withameliorationof deficiency and prevention of neurological sequelae.
HypeNitaminosis E
Relatively large amounts of vitamin E, in range of 400 to 800 mg tocopherol, have been taken daily by adults for months to years without causing any apparent harm. Occasionally, muscle weakness, fatigue, nausea and diarrhea are reported in persons ingesting 800-3200 mg/ day. Vitamin E intoxication, at dosages exceeding 1,000 mg/day, results in antagonism to vitamin K action and enhanced effect of oral coumarin anticoagulants,with hemorrhage.
VitaminK
Vitamin K is a generic term for derivatives of 2-methyl-l, 4-naphthoquinone with procoagulant activity. This is an essential vitamin, since the 1,4-naphthoquinone moiety is not synthesized in animal cells. The natural forms are substituted in position 3with an alkyl side chain. Vitamin Kl (phylloquinone) has a phytol side chain in position 3 and is the homolog of vitamin K in plants. Vitamin K2 (menaquinone), with an isopropyl side chain, is synthe sized by bacteria in the gut.
Absorption and metabolism The absorption of phyllo quinone and menaquinone require bile and pancreatic juice. Dietary vitamin K is absorbed in the small bowel, incorporated into chylomicrons and delivered to the circulation via the lymph. The liver is the primary site of action of vitamin K. The total body pool of vitamin K is small, 80% being in the liver.
Physiological function The main role of vitamin K is as a cofactor in post-translational carboxylation of glutarnic acid to formglutamatein the liver. Vitamin K carboxylates glutamic acids of translation products of vitamin K dependent proteins, to produce y-carboxyglutamates. Factors II (prothrombin), VII, IX and X are procoagulant proenzymes whereas proteins C and S are anticoagulant proenzymes. The function of these proteins is to facilitate the chelation of calcium ions to glutamate and platelet phosphatide,whichisessentialfor thecoagulationcascade to operate.

Nutritional requirements Vitamin K requirements are met by combination of dietary intake and microbiological biosynthesis in the intestines. The vitamin K dependent coagulation factors are <30% of normal at birth in full terminfants andeven lower in premature newborns. Vita min K requirement for the normalization of prothrombin and other factor in newborns is 3 to 5 µg/day.Since breast milk contains only 2 µg/L phylloquinone, breastfed infants should receive additional vitamin K to prevent hemorrhagic disease of the newborn. The requirement increases from 5 µg/day at birth to 10 µg/day at 2 yr and 10 to 30 µg/day in older children.
Sources Green leafy vegetables are rich in phylloquinone, animal foods are intermediate and cereals low in the vitamin. Bacterial gut flora is capable of synthesizing the vitamin, sufficient to meet daily needs.
Vitamin K Deficiency
Most natural foods have high vitamin K content, relative to its requirement. Thus, primary deficiency is very rare. Deficiency can however result from elimination of intestinal bacterial flora, exclusive parenteral alimentation with no added vitamin K, fat malabsorption, biliary obstruction, cystic fibrosis and short bowel syndrome.
Hemorrhagic disease of the newborn is a syndrome of systemic bleeding and ecchymoses appearing in the first week of life, predominating in breastfed infants. Because of routine administration of prophylactic vitamin K at birth, most cases of hemorrhagic disease of the newborn are of late onset (after 2 weeks of life) and are associated with a variety of conditions such as antibiotic therapy, cholestasis, maternal use of antagonist drugs (primidone, warfarin, diphenylhydantoin), low dietary intake and fat malabsorption. Confirmation ofthe diagnosis depends on a rapid therapeutic response to administration of vitamin K intramuscularly. Healthy newborns should receive vitamin K at a dose of 0.5-1.0 mg IM to prevent hemorrhagic disease. Parenterally fed infants and children should receive 1 mg vitamin K once weekly.
WATER SOLUBLE VITAMINS
Thiamine (Vitamin B1)
Requirements The recommended daily allowance is 0.4 mg/1000 kcal of carbohydrate intake.
Dietary sources These include unrefined or fortified cereal grains, enriched bakery products, organ meats (liver, kidney) and legumes. Thiamine is sensitive to heat, sulfites, pasteurization and sterilization; freezing results in little loss. Thiamine content of human milk is relatively low (16 µg/ml) compared to cow milk (40-50 µg/ml).
The vitamin is absorbed chiefly from the jejunum throughasodiumdependent mechanism;ethanolinhibits absorption.Althoughthiamine deficiencyduringpregnancy is described, the fetus appears protected by active
Micronutrients in Health and Disease -
placentaltransport;vitaminlevels in cordblood are higher than in maternal blood.
Biologic action Thiamine pyrophosphate is involved in several enzymatic steps of carbohydrate metabolism.It is a cofactor for oxidative decarboxylation of pyruvate to form acetyl-CoA, a step catalyzed by the pyruvate dehy drogenase complex. This pathway is not directly involved in carbohydrate metabolism, but is the major source of five carbon compounds for nucleic acid synthesis and NADPH forfatty acid synthesis.The transketolase reaction is affected rapidly in thiamine deficiency, and its rate in erythrocytes is used as an index of thiamine status.
Deficiency Thiaminedeficiencyor beriberi affects people who consume diets based on polished rice, when the intake is below 1 mg/day. The classic signs of beriberi appear after prolonged periods of low thiamine intake. Three forms of beriberi are described: dry, wet and acute. The dry and wet (edematous) forms are different mani festations of a polyneuritis. The dry form has no edema and typically includes severe muscle wasting and cardio megaly.Wetberiberiis characterized by peripheral edema, ocular paralysis, ataxia and mental impairment. The pathogenesis of edema in wet beriberi is unclear. Infantile beriberi may be more subtle than that found in adults. It occurs in breastfed infants of thiamine-deficient mothers (who may not have signs of beriberi), or with very low thiamine intake. The clinical picture is dominated by cardiomegaly, cyanosis, dyspneaandaphonia. Thedisease mayresult in death aftera few weeks, in the infantile form. Diagnosis Thiamine deficiency may be suspected in all cases of malnutrition. The diagnosis is confirmed by measurement of 24 hr urinary thiamine excretion, which in children is 40-100 µg/day; values below 15 µg/day are deficient. Diagnosis of deficiency can also be based on the response of red cell transketolase to the addition of thiamine in vitro. Erythrocytesfromdeficientpersons have a greater response to thiamine pyrophosphate addition than do those of normal controls. An increase in trans ketolase activity of less than 15% is normal, 15-25% is mild deficiency and over 25% are severely deficient.
Treatment Treatment with thiamine leads to resolution of neurologic and cardiac symptoms within 24-48 hr. Treatment of patients with mild beriberi with thiamine (5 mg/day) is satisfactory. Severely ill children should receive 10 mg intravenously twice daily. In management of fulminant heart disease, higher doses with vigorous treatment of congestive heart failure are necessary.
Riboflavin (Vitamin 82)
Riboflavin is a flavoprotein, widely distributed in plants. Meat, poultry fish and dairy products and broccoli, spinach and asparagus are good sources. Riboflavin is resistant to oxidation and to heat and is not destroyed by pasteurization. Human milk contains 40-70 µg/100 kcal

e |
i |
i |
i |
s ------------ |
- |
|
ss e_n_ta r ePd atr_-c------------------ |
|
- |
||||
|
|
|||||
of riboflavin and cow milk 250 µg/100 kcal. Maternal riboflavin supplementation may be used to increase its concentration in breast milk. Riboflavin is efficiently absorbed from the small intestine by site-specific, saturable, specialized transport. Biliary obstruction, high dietary fiber and antacids decrease its bioavailability.
Riboflavin is a constituent of two coenzymes involved in oxidation-reduction reactions: flavin adenine dinucle otide and flavin mononucleotide. A number of redox enzymes, including glutathione reductase and xanthine oxidase,requireflavincoenzymes.Theenzymescatalyzing the synthesis of niacin from tryptophan and the conversion of pyridoxal phosphate to an active coenzyme are flavin dependent and link riboflavin with these two vitamins. Riboflavin deficiency affects fatty acid synthesis and decrease in plasma levels of linoleic and linolenic acids. By impairing conversion of phosphorylated vitamin B6 to its coenzyme, it affects reactions involved in amino acid metabolism requiring this vitamin.
Riboflavin requirements The recommended daily intake is 0.4 mg/1000 kcal for infants and 0.8-1.2 mg/1000 kcal for children.
Deficiency Riboflavindeficiency occurs frominadequate intake or malabsorption. It takes 1-2 months to develop and is associated with other deficiencies. Features of ariboflavinosis are photophobia, glossitis, angular stomatitis, seborrheic dermatitis, corneal vascularization and cataracts. Nonspecific symptoms include anorexia, weight loss, weakness, dizziness and confusion.
Diagnosis of deficiency Diagnosis should beconsidered with a history of dietary deficiency and clinical mani festations. A reliable indicator of riboflavin status is the daily losses of the vitamin; urinary excretion of less than 10% of intake over 24 hr isindicativeofdeficiency. Activity of glutathione reductase inerythrocytes gives a functional index of flavin coenzyme activity; cofactor-induced increase of 20% above the basal levelindicates deficiency.
Treatment Children are treated with 1 mg riboflavin threetimesdaily for severalweeks; infants respond to 0.5 mg twice daily. Therapeutic doses of vitamin help in improving corneal lesions rapidly.
Niacin (Vitamin B3)
Nicotinic acid and nicotinamide, biologically equivalent vitamins, are both referred to as niacin. Biosynthesis of this vitamin occurs in all organisms; the conversion ratio of tryptophan to nicotinic acid is 60:1, making it possible for large amounts of tryptophan to meet niacin needs.
Sources Niacin is distributed in plants and in animal foods,onlyinpyridinenucleotideform.Milk,cereals,leafy vegetable,fish,coffeeandteaaregoodsources.Thevitamin is resistant to heating. Human milk contains 30 mg/100 kcalofniacincomparedwith0.12mg/100kcalincowmilk.
Absorption and metabolism Niacin is absorbed in the
proximal small bowel and incorporated into nicotinamide adenine dinucleotide (NAD) and its phosphate (NADP). Excess niacin is methylated in the liver, forming N1 - methylnicotinamide. This compound and its oxidation product, 2-pyridone, are the two major niacin metabolites found in urine.
Niacin requirements Requirements are expressed in terms of niacin equivalents (NE) One NE equals 1 mg of
niacin or 60 mg of tryptophan. RDA for niacin is related to dietary energy intake; the recommended intake is 6.4 to 8 NE/1000 kcal, human milk provides about 8 NE/ 1000 kcal. High doses of nicotinic acid (but not nicotina mide) reduce serum cholesterol and triglyceride levels in humans. A number of side effects, including skin flares, hyperuricemia, hyperglycemia and abnormal tests ofliver function have been described.
Niacin deficiency Niacin deficiency leads to pellagra, the pathognomonicskin change. It was originally thought to be caused by a factor deficient in maize, subsequently identified as nicotinic acid. Clinical features of deficiency are chronic, relapsing and popularly characterized by three Ds: dermatitis, diarrhea and dementia. The cuta neous lesions consist of a pigmented rash aggravated by sunlight. More acute cases may progress to vesiculation, ulceration and secondary infection. Classically, the ery thema progresses to roughening and keratosis with scaling; a characteristic red tongue is seen. While neuro logic features may appear without skin manifestations, they usually followthe skinlesions. Neurologic symptoms include apathy, headache and loss of memory. In most chronic forms, posterolateral cord degeneration and peripheral nerve lesions areseen.Onlyin the most severe and chronic cases, the neurologic lesions persist after adequate treatment with niacin.
Diagnosis of niacin deficiency The diagnosis is suspec
tedonhistoryofinadequatediet,INHtreatmentorchronic alcohol ingestion when typicalmanifestationsare present. Determination of urinary excretion of N1-methylnico tinamide is most helpful; normal 24 hr excretion is bet ween 4 and 6 mg, values below 3 mg indicate deficiency. In pellagra these values are usuallybetween 0.5 to 0.8 mg/ day.
Treatment of pellagra The daily dose for treatment is about 10 times the recommended dietary intake. Oral treatment with nicotinamide is preferred to nicotinic acid to avoid unpleasant side effects. Parenteral therapy is considered when gastrointestinal absorption is deficient. Prevention of pellagra is achieved by an adequate protein diet containing tryptophan and niacin rich foods.
Pyridoxine (Vitamin B6)
Pyridoxine aids in food assimilation and protein and essential fatty acid metabolism. It activates many enzyme

systems and is involved in the production of antibodies against bacterial diseases. It is linked to cardiovascular health by decreasing the formation of homocysteine. It is also required for absorption of vitamin B12 and for production of monoamine neurotransmitters serotonin, dopamine, noradrenaline and adrenaline. Lack of pyri doxine may cause anemia, neuropathy, seizures, skin problems andmouth sores. Pyridoxine is given 10-50 mg/ day to patients on INH (isoniazid) to prevent peripheral neuropathy and other neurologic effects.
Vitamin B6 is absorbed mainly in the jejunum. Although colonic bacteria synthesize B6, it isnotabsorbed to significant extent. Small quantities of the vitamin are stored in the body. Rich sources of vitamin B6 include yeast,sunflowerseeds,wheatgerm, soyabeansandwalnuts.
Cobalamin (Vitamin B12)
Cyanocobalamin is the most common commercially availableformof vitaminB12,buttheactive formsin tissues are methylcobalamin and 5'-deoxyadenosyl cobalamin. Methylcobalamin isa coenzymein thereactionthattransfers a methyl group to homocysteine to form methionine. The methyl group donor for this reaction ismethylfolate, which is then regenerated to its active form, tetra-hydrofolate. VitaminB12 deficiencyblocks thisreaction,trappingfolate into the inactive methyl form, thus favoring the develop ment of folate deficiency. The other coenzyme, 5'- deoxyadenosyl cobalamin, helps in formation of succinyl CoA, an essential step in catabolism of valine, isoleucine and other amino acids through the Kreb cycle.
Sources Vitamin B12 is only produced by micro organisms. Animals contain cobalamin either ingested with micro-organisms or produced by bacteria in the upper segments of the intestine. Highest concentrations arein organs such as liver, kidney, heart andmuscle meat; clams and oysters are excellent sources.
Absorption and metabolism A specific receptor mediated process in the ileum involves a glycoprotein, intrinsic factor. Absence of this gastric protein results in pernicious anemia, an inability to absorb ingested vitamin B12.Passivediffusionaccountsfora fractionoftotalabsorp tion, but has implications for management of pernicious anemia with megadoses of vitamin.Cobalamin undergoes enterohepatic recirculation; this process accounts for a long half-life of the vitamin. Vitamin B12 is transported in plasma bound to transcobalamin II. The average total body pool in an adult is enough to sustain daily vitamin B12 needs for several years.
Requirements The recommended intake of vitamin B12 for infants is 0.3 µg/day. Older children should receive 0.5-1.5 µg/day and adolescents 2.0 µg/day. These needs are met by any mixed diet.
Deficiency Most deficiencies are caused by impaired absorption, due to the deficiency of the intrinsic factor, or
Micronutrients in Health and Disease
intestinal or liver disease and they are more common in adults and elderly persons. True dietary vitamin B12 deficiency occurs in persons who follow strict diets containing no animal orfishproducts. VitaminB12 status is assessed by measurement of serum cobalamin levels, with values below 1.1 pmol/1 indicative of negative vitamin B12 balance. Plasma levels of methylmalonic acid and homocysteine are increased because of block in vitamin B12-dependent steps of metabolism. Methyl malonic aciduria may also occur but is a less consistent finding. Since development of clinical vitamin B12 deficiency takes a long time, itisrarein infants. Exclusively breastfed infants of strict vegetarian mothers, however, may be at risk ofdeficiency,manifested bymethylmalonic aciduria and megaloblastic anemia. Other features of deficiency are neutrophil hypersegmentation, megalo blastic anemia and thrombocytopenia.
A specific feature of B12 deficiency is a diffuse and progressive demyelination, which begins in peripheral nerves and progresses to involve the posterior and lateral columns of the spinal cord and central nervous system. These lesions are possibly due to a generalized methyl group deficiency in the nervoussystem and perhaps also to toxic accumulation of homocysteine.
Diagnosis of deficiency The anemia is macrocytic and nucleated RBC showing megaloblastic morphology may be seen in blood. Levels of red cell folate are low; serum LOH levels are elevated.
Treatment Deficiencyis treated with parenteral adminis trations of vitamin B12 (1 mg). Reticulocytosis is seen within 2-4 days. Patents with neurologic involvement require day therapy for 2 weeks.
Folic Acid (Pteroylglutamic Acid)
Folic acid is the parent compound of a group of naturally occurring, structurally related compounds known as the folates. Folic acid is essential for normal growth and maintenance of cells, sinceit acts as a coenzymefornormal DNA and RNA synthesis. Folate is vital for multiplication of cells within the fetus. A deficiency therefore affects nor mal cell division and protein synthesis, impairing growth. Folic acid, with vitamin B12 converts homocysteine to methionine, thereby reducing blood levels of homo cysteine and lowering risks of heart disease. It also main tains integrity of thecentralnervous system and intestinal tract function and is involved in production of neurotransmitters suchasserotonin. Leafyvegetables such as spinach, turnip greens, lettuces, dried beans and peas, fortified cereal products, sunflower seeds and certain fruits and vegetables are rich sources.
Requirements Therecommendeddailyallowanceof folic acidvaries from 25 mg in infancy to 200mg by adolescence (Table 7.1). Deficiency is corrected using folic acid at a dose of 0.5-1 mg/day orally for 3-4 weeks.

i |
i i |
s |
|
_____________ |
__E_se_sna_tt__P_daetr |
c |
______________ |
_ |
|
|
||||
Deficiency of folate limits cell division and protein synthesis and affects normal growth and repair of tissues. The tissues that have the fastest rate of cell replacement are affected first. Erythropoiesis is hindered, resulting in megaloblasticanemia; other symptoms are impaired brain and nerve functions, especially memory problems. Maternal deficiency of folate, during pregnancy, is implicated in neural tube defects. Periconceptional folate supplements, begun 1 month before conception and continued for 3 months after, is recommended to reduce the risk.
Biotin
Biotin is acoenzyme for carboxylationreactions. Four such reactions require biotin as a cofactor: (a) acetyl-CoA carboxylase synthesizes malonyl-CoA, the initial step in fattyacid synthesis; (b) pyruvate carboxylaseforms oxalo acetate, a key intermediate of the Krebs cycle; (c) pro pionyl-CoA carboxylase forms methylmalonyl-CoA, permitting the entry to the Krebs cycle of carbons derived from branched chain amino acids, fatty acids and cholesterol; (d) methylcrotonyl-CoA carboxylase parti cipates in the catabolism of leucine.
Biotin deficiency has been observed in individuals who consume large number of raw eggs (rich in avidin) for several months. The avidin is not hydrolyzed by gastro intestinal enzymes; it binds biotin and prevents its absorption. Cooking of eggs destroys avidin. Clinical features of biotin deficiency include anorexia, vomiting, dry scaly dermatitis, glossitis and hypercholesterolemia. Longterm parenteral alimentation without biotin can also lead to deficiency in pediatric and adult patient. Multiple carboxylase deficiency is a genetic metabolite disorder affecting the activity of carboxylase synthetase, which catalyzes the transfer of biotin to the apocarboxylase moiety. This condition responds to large doses of biotin. Another genetic defect affects the activity of biotinidase, an enzyme involved in the recycling of biotin.
Dietary sources of biotin include liver, egg yolk, milk, yeast extracts and meat. Requirements are difficult to determine, since biotin is produced by gastrointestinal flora. Recommendations are 0.15 mg biotin in the multivitamin supplements for infants and children. For treatment of biotin deficiency, oral administration of 2-5 mg daily for 2 to 3 weeks is recommended for mild cases. A parenteral biotin dose of 200 µg daily for 2 to 5 days can be used in more severe cases.
Pontothenic Acid
Pantothenic acid (vitamin BS) is present in virtually all naturally occurringfoods andis also synthesized bymicro organisms from pantoic acid and -alanine. Pantothenate is absorbed in the proximal small intestine; in the liver it becomesanintegralpart of coenzymeA,whichis essential for acyl transfer reactions of fatty acid, steroids and
cholesterol synthesis, as well as for the metabolism of pyruvate and a-ketoglutarate. Isolated pantothenate deficiency israreand includes burning feet, insomnia and gastrointestinal symptoms. In cases of extreme mal nutrition, such a deficiency coexists with other vitamin deficiencies. The suggested daily intake is 2-3 mg for infants and 3-5 mg for children.
Vitamin C
Vitamin C (ascorbic acid), structurally related to glucose, has a striking capacity for reversible oxidation reduction. Humans and other primates do not synthesize vitamin C.
Sources Dietarysourcesincludevegetables (cauliflower, broccoli, cabbage) and fruits (berries, citrus). Much of vitamin C may be lost in cooking, but is stable in canned and frozen foods. Vitamin C in human and cow milk ranges from 5 to 15 mg/100 kcal and 0.2-2.0 mg/100 kcal respectively. Daily requirements are 30-40 mg for infants and 40-70 mg for children.
Absorption and metabolism Ascorbic acid is absorbed by anactive,sodium-dependentprocessin the upper small intestine. The vitamin circulates in plasma in its free, anionicform, reaching high concentrations in adrenal and pituitary glands and in leukocytes. Vitamin C appears unchanged in the urine when renal threshold is exceeded. Biologic action Vitamin C functions as a strong reducing agent or in electron transport within biological systems. Ascorbicacidisessentialfor normal function of leukocytes, fibroblasts, osteoblasts and microsomes and participates in metabolism of carnitine, serotonin and folate. Ascorbic acid affects the immune response, detoxification, collagen synthesis and wound healing.
Deficiency Prolonged vitamin C deficiency results in scurvy. Itusuallyoccurs in those who aredeprivedof citrus fruits, fresh vegetables or vitamins for some cultural or geographic reasons. In infancy, features of scurvy are anorexia, diarrhea, pallor, irritability and increased susceptibility toinfections. Subperiosteal hemorrhages and long bone tenderness (pseudoparalysis of lower extre mities) can occur; radiologic abnormalities are frequent. In older children, hemorrhagic signs predominate, with bleeding from gums, conjunctiva and intestinal tract.
Diagnosis of scurvy The diagnosis is made by presence ofcharacteristicphysicalfindingsand historyof inadequate dietary intake of vitamin C. X-rays of long bones show a ground glass appearance with thinning of cortex and sharply outlined epiphyseal ends. The finding of a zone of rarefaction under the white line of metaphysis, as a linear break in the bone runningproximal andparallel to thewhite line, is diagnostic in infantile scurvy. The lateral part of the rarefaction appears as a triangular defect. There may be spurs at the level of the white line. Other characteristic

findings include the white line of Frenkel, an irregular thickened line at the metaphyses representing calcified cartilage at the zone of calcification and white rings surrounding the epiphyseal centers of ossification. Vitamin C therapyoften results in dramatic improvement within 24-48 hr.
Therapy for scurvy Therapy with 100-200 mg of vitamin C orally or parenterally results in prompt and rapid improvement in symptoms. Daily intake of 100 ml of orange juice or tomato pulp has the same effect.
MINERALS AND TRACE ELEMENTS
Calcium
Calcium is the most abundant mineral in the body and is located primarily (98%) in bone. Calcium is essential for the coagulation cascade, nerve conduction and muscle stimulation. Intestinal absorption of calcium varies inversely with intake and is regulated by 1,25(0H)D3, which controls the synthesis of calcium-binding protein at the brush border. In the presence of vitamin D, calcium absorption can adapt to a wide range of dietary calcium intakes, varying from 10 to 80% of available calcium. Calcium absorption also depends on the interaction of calcium with other dietary constituents, including fiber, phytate, oxalate, fat and lactose.
The main sources of calcium for infants are milk and dairyproducts,with smaller amounts derived from grains and fruits once solid foods are introduced. Children consuming strict vegetarian diets may develop calcium deficiency, either alone or in combination with vitamin D deficiency. Strict vegetarian diets may provide as little as 250 mg of calciumper day and include generous amounts of substances that inhibit calcium absorption, such as fiber and phytates. Secondary calcium deficiency may develop in association with steatorrhea, chronic malabsorption syndromes, or intestinal or renalabnormalitiesof calcium metabolism.
Children aged 1 to 10 yr require an intake of 500 to 800 mg per day. Duringthepubertalgrowth spurt calcium requirements are as high as 1000 to 1200 mg per day. Pregnant and lactating women require 400 mg per day. Calcium deficiency may cause tetany characterized·by muscle cramps, numbness and tingling in limbs. Rickets and osteoporosis may occur with chronic deficiency.
Magnesium
Magnesium is essential for bioenergetic reactions con trolling fuel oxidation, membrane transport and signal transmission contributing to the action of more than 300 enzymes.Over 80% of thetotal body magnesiumis in bone and skeletal muscle. Rich sources of magnesium include legumes, nuts, bananas and whole grains. Magnesium is absorbed efficiently by the intestine and regulation of its balance depends onrenal tubular reabsorption. Deficiency
Micronutrients in Health and Disease -
isusuallysecondarytointestinalmalabsorption,excessive gastrointestinal losses through fistulae or continuous suction or renal disease affecting tubular reabsorption. Clinical manifestations of magnesium deficiency include irritability,tetanyandhypo orhyper-reflexia. Magnesium requirements in the first 6 months range between 40 and 50 mg/day; 60 mg/day for 6-12 months and approxi mately 200 mg/day for older children.
Trace Element Deficiencies
Eleven 'major' elements constitute 99% of human body weight. These essential-for-life elements are hydrogen, carbon, nitrogen, oxygen, sodium, potassium, chlorine, calcium,phosphorus,sulfurand magnesium. In addition, the body is composed of numerous "trace" elements. The term trace elements comprise an increasing number of compoundswithprovenorputativeessentialityforhuman nutrition.Each ofthesecontributesless than 0.01% oftotal bodyweight. Their majorfunctions arerelated to enzyme systems where they act either as cofactor for metal-ion activated enzymes or as specific constituents of metallo enzymes. This section provides an overview of all trace elements of established importance in human nutrition.
Zinc
Functions Zincis acomponentofover100metalloenzymes and participates in many biological processes. As a component of zinc finger proteins, zinc regulates gene transcription andparticipatesin nucleic acid metabolism, protein synthesisand thereby,cellular growth. Thyrnidine kinase, DNA polymerase and RNA polymerase are reported to be zinc dependent enzymes.
Absorption and metabolism Zinc is absorbed through out the small intestinebya process of facilitated diffusion. Most absorbed zinc is taken up temporarily by the liver, which plays a central role in zinc metabolism. Absorbed zinc is transported in the portal system attached to albu min or transferrin. In the systemic circulation, the major fraction of plasma zinc is loosely bound to albumin. Almost 90% of total body zinc is localized in bone and skeletal muscle. Zinc status is regulated both at the absorptive step and by intestinal re-excretion. The major excretory route for endogenous zinc is via the feces.
Deficiency Zinc deficiency is usually seen as a part of malnutrition or malabsorption syndromes,causedbylow dietary intake orintestinaldisease. Severe zinc deficiency syndromes have occurred in patients on prolonged intravenous feeding without adequate trace element supplements.Poorphysicalgrowth is an important feature of zincdepletion inpreschooland school-age children and its supplementation results in accelerate growth in adolescents with intestinal malabsorption and sickle cell disease. Delayed sexual maturation and hypogonadism is also a prominent feature of zinc deficiency in adole scents. Other features include anemia, anorexia, diarrhea,

___E_s_s_e_n_ t_ia_i_P_e_d_ i_a _trics_________________________________
hair loss, dermatitis, impaired immune function, poor wound healing and skeletal abnormalities.
Acrodermatitis enteropathica is an autosomal recessive syndrome of severe zinc deficiency, caused by defective intestinal absorption due to defect in intestinal zinc transporter protein. Presentation is in early infancy, with vesicobullous, dry, scaly oreczematousskinlesionschiefly involving the perioral, perinea! and acral areas. Alopecia and eye changes, such as conjunctivitis, blepharitis and photophobia, may be present. Chronic diarrhea, growth retardation, stomatitis, irritability and delayed wound healingare otherfindings.Catchupgrowthand resolution of symptoms is noted following oral zinc therapy.
The diagnosis of zinc deficiency is often derived from the combination of dietary history of chronic low zinc intake or excessive intestinal losses, presence of clinical features compatible with deficiency and low levels of zinc in plasma or hair.
Requirementandtreatmentof deficiency The normal requirements for children range between 3.5 and 5.0 mg per day. Acquired zinc deficiency states can be treated with 0.5 to 1.0 mg elemental zinc/kg/day for several weeks or months. One mg of elemental zinc is available from 4.5 mg zinc sulfate or 3 mg zinc acetate. Intravenous requirements for patients maintained on prolonged intravenous feeding approximate 50 µg of elemental zinc/kg body weight/day. These requirements can be considerably higher in the presence of excessive zinc losses.Zinctherapyshouldbemonitoredwithplasmazinc and copper concentrations as excessive zinc therapy can lead to a copper deficiency syndrome.
Copper
Copper is acomponent ofseveralmetalloenzymesrequired for oxidative metabolism. Ceruloplasrnin, a glycoprotein that contains eight copper atoms per molecule, accounts for 95% of the ion in blood.
Absorption and metabolism Approximately 40% of ingestedcopper isabsorbedinstomachandsmall intestine, from where it is transported to the liver bound to albumin andutilizedbyhepatocytesfor synthesis of ceruloplasmin that is subsequently releasedinto the systemic circulation. Approximately one-third of fecal copper is contributedby bileacids,therestisunabsorbedcopperandfromepithelial desquamation; urinary excretion is minimal.
Sources The richest sources are meats, liver, seafood, nuts and seeds. Additional copper may enter the food chain through pesticides and contamination of water by pipes and cooking utensils.
Deficiency Primary dietary deficiency is infrequent. Secondary deficiency may develop in malabsorption syndromes, liver disease, peritoneal dialysis and other conditions causing excessive copper losses. Features of deficiency are rnicrocytic, hypochrornicanemiaunrespon-
sive to iron therapy, neutropenia and osteoporosis. Copper deficiency decreasesthelifespan oftheerythrocyte and impairs mobilization of stored iron from liver and bone marrow. Skeletallesions include periosteal elevation, changes in metaphyses of long bones, and rarely sub metaphyseal fractures, flaring of the anterior ribs and spontaneous fractures of the ribs. Infants show pallor, depigmentation of skin and hair, prominent dilated superficial veins, lesions resembling seborrheic dermatitis, anorexia, diarrhea and failure to thrive.
Copper transport is disrupted in two human diseases: Wilson disease and Menkes disease. Both have defects in copper transporting membrane proteins. Central nervous system manifestations include hypotonia, psychomotor retardation andapneicepisodes.In the Menkessteely-hair syndrome there is severe neurological degeneration lead ing to a fatal outcome by early childhood. Laboratory findings include hypocuprernia, lowplasma ceruloplasrnin, neutropenia and anemia.
Selenium
Selenium is a constituent of glutathione peroxidase, an antioxidant in red blood cells and other tissues. Glutathione peroxidase scavenges free hydroperoxides generated during fatty acid oxidation, thus protecting the cell from damage due to free radical formation. Severe deficiency is the major cause of Keshan disease, which presents as a cardiomyopathy in young children. Skeletal myopathies have also been reported. Mild deficiency is associated with macrocytosis and loss of hair pigment.
Chromium
Glucose intolerance, which complicates malnutrition in young children, has been attributed in part to chromium deficiency. Chromium acts in glucose homeostasis by potentiating insulin action, possibly by facilitating binding to its receptor. Symptoms of chromium deficiency are usually in the setting of total parenteral alimentation and include glucose intolerance, peripheral neuropathy and evidence of disturbed nitrogen and lipid metabolism.
Iodine
The term iodine deficiency disorders (IDD) refers to the effects of iodine deficiency in a population that can be preventedbyensuringanadequateintakeofiodine. Iodine deficiency disorders (IDD) jeopardize children's mental health and often their very survival. Serious iodine deficiency during pregnancy can result in stillbirth, spontaneous abortion and congenital abnormalities such ascretinism,anirreversibleformofmental retardation that affectspeople livingin iodine-deficient areas. However, of fargreatersignificanceisthelessvisiblementalimpairment that reduces intellectual capacity at home, in school and at work. The effects of iodine deficiency on growth and development are summarized in Table 7.3.
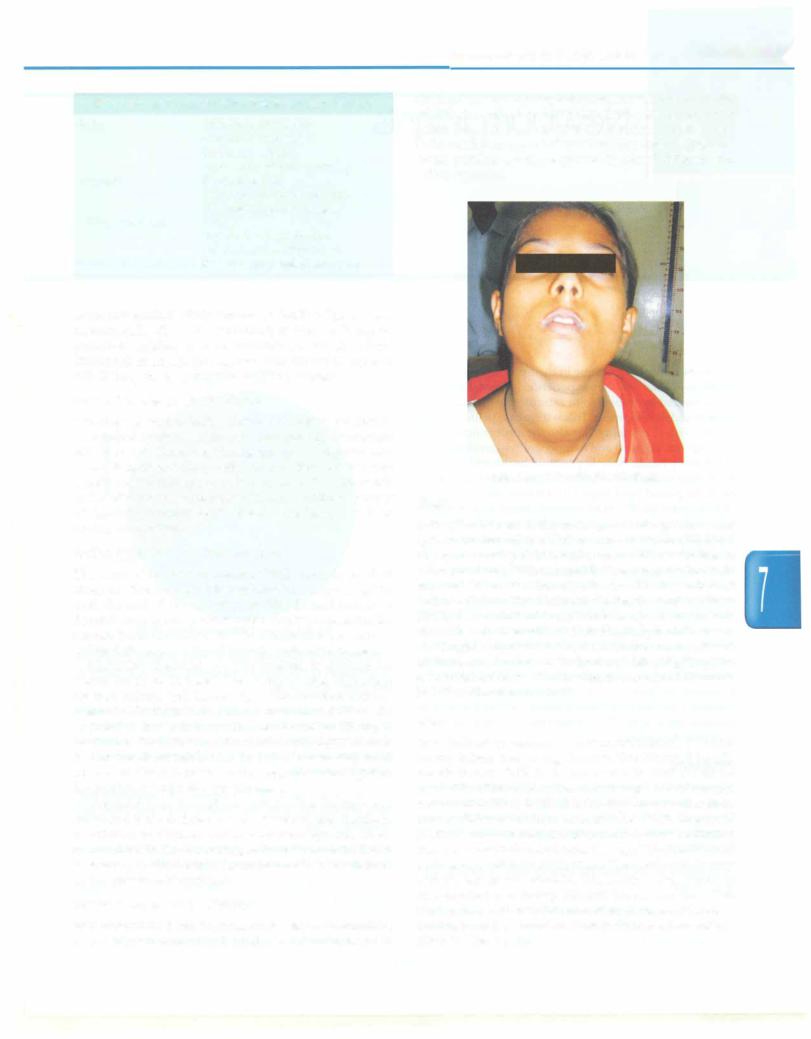
Table 7.3: Spectrum of iodine deficiency disorders IDD
Fetus |
Abortions, stillbirths |
|
Congenital anomalies |
|
Endemic cretinism |
|
Increased perinatal mortality |
Neonate |
Neonatal goiter |
|
Endemic mental retardation |
|
Neonatal hypothyroidism |
Child, adolescent |
Goiter |
|
Impaired mental function |
|
Subclinical hypothyroidism |
|
Retarded physical development |
Recommended daily intake of Iodine The recom mended daily allowance of iodine is as follows: 90 mg for preschool children (0 to 59 months); 120 mg for school children (6 to 12 yr); 150 mg for adults (above 12 yr); and 200-250 mg for pregnant and lactating women.
Iodine Deficiency in the Fetus
The consequence of iodine deficiency during pregnancy is impaired synthesis of thyroid hormones by the mother and the fetus. Since the physiologic role of thyroid hor mones is to ensure the coordination of different develop mental events through specific effects on the rate of cell differentiation andgene expression, an insufficient supply of thyroid hormones to the developing brain results in mental retardation.
Iodine Deficiency in the Neonate
The brain of the human infant at birth has only reached aboutone-thirdofits fullsizeandcontinuestogrowrapidly until the end of the second year. The thyroid hormone, dependenton an adequate supply of iodine, isessentialfor normal brain development. The continuing presence of iodine deficiency is a threat to early brain development.
Neonatal chemical hypothyroidism is defined by serum levels of T4 lesser than 3 mg/dl and TSH grea ter than 100mU/ml. In severely iodine deficient environ ments in Northern India, where more than 50% of the population has urinary iodine levels below 25 mg/g creatinine, the incidence of neonatal hypothyroidism is 75-115 per thousand births. In Delhi, where only mild iodine deficiency is present with low prevalence of goiter, the incidence drops to 6 per thousand.
Neonatal hypothyroidism persists into infancy and childhood if the deficiency is not corrected and results in retardation of physical and mental development. These observations indicate a much greater risk of mental defect in severely iodine-deficient populations than is indicated by the presence of cretinism.
Iodine Deficiency in Children
Moderateiodinedeficiencyisassociatedwithabnormalities
in psychoneuromotor and intellectual development of
Micronutrients in Health and Disease -
childrenwhoareclinicallyeuthyroid, butwhodonotexhibit otherfeaturesofendemiccretinism.Somepatientsmayshow goiter (Fig. 7.7) (see Chapter 17). Studies in moderately iodine-deficient areas indicate that fine motor skills and visual problem solving improved in school children after iodine repletion.
Fig. 7.7: A 14-yr-old girl with goiter
Therapy
Iodization of salt is the most practical option. Other optionsforcorrection of IDDareadministration of iodized oil capsules every 6-10 months, direct administration of iodine solutions, such as Lugol iodine at regular intervals and iodization of water supplies by direct addition of iodine solution. The National Goiter Control Program
(1962) for control of iodine deficiency disorders was started by establishment ofsalt iodination plants to ensure an adequate supply of iodized salt in the country. Based on an assumption of a mean intake of salt of 5 g/day, the recommended level of iodination is one part of iodine in 25,000 to 50,000 parts of salt.
Iron
Iron deficiency remains a major nutritional problem among infants and young children. The National Family Health Survey (NFHS) II, conducted in 1998-99, docu mented that 74%childrenbetweenthe ages of 6-35 months were anemic. The NFHS III (2005-06) shows similar data. Iron deficiency anemia is associated with impaired performance in mental and physical functions, including physical coordination andcapacity,cognitiveabilities, and social and emotional development. Thepreciseeffectsvary with the age groups studied. The health consequences of iron deficiency in young children are serious and often irreversible. A description of clinical features, diagnosis, treatment and prevention of iron deficiency anemia is given in Chapter 12.

Newborn Infants
Newborn infants are unique in their physiology and the health problems that they experience. Neonatal period is characterized by transition to extrauterine life and rapid growth anddevelopment.This is thephase in life with the greatest risk of mortality. It is also the most critical period for longterm physical and neurocognitive development.
Newborn health is the key to child health and survival. More than under half of under 5 child deaths occur in the neonatal period (see Chapter 1). Preterm birth complica tionsaccount for 35% of all neonatal deaths and constitute the most important cause of neonatal mortality (Fig. 8.1). Bacterial infections (sepsis, pneumonia and diarrhoea) contribute to 33% of neonatal deaths. Other causes of neonatalmortalityarebirthasphyxia(20%)andcongenital malformations (9%). Almost three-fourths of all neonatal deaths occur among the low birth weight newborns. Of all theneonataldeaths,about40%occurwithinfirst24hr, half within 72 hr and three fourths within one week of birth. Predominantcausesofdeath inthefirstweekoflifeinclude birth asphyxia and preterm birth complications. Health of themotherandcareduring pregnancyandatchildbirthhas profound influence on neonatal outcome. As noted in Chapter1, decline in neonatal mortality iscritical toachieve nationalhealthgoals.Thestagnantearlyneonatalmortality is a cause for concern.
Definitions
Neonatal period. From birth to under four weeks (<28 days) of age. An infant is called a neonate during this phase. First week of life (<7 days or <168 hr) is known as early neonatal period. Late neonatal period extends from 7th to <28th day.
Postneonatal period. Period of infancy from 28 days to <365 days of life.
Weeks ofgestation refer to completed weeks of gestation, e.g. 36 weeks gestation, refer to range of gestation from 36 weeks O day to 36 weeks and 6 days.
Ramesh Agarwal, Vinod K Paul, Ashok K Deorari
Fig. 8.1: Causes of neonatal deaths (2010)
Perinatal period. Perinatal period extends from 22nd week of gestation (L154 days or weighing L500 g at birth) to less than 7 days of life.
Live birth. A product of conception, irrespective of weight or gestational age, that, after separation from the mother, shows any evidence of life such as breathing, heart-beat, pulsation of umbilical cord or definite movement of voluntary muscles.
Fetal death. A fetal death is a product of conception that, after separation from the mother, does not show any evidence of life.
Still-birth. Fetal death at a gestational age of 22 weeks or more or weighing more than 500 g at birth.
Term neonate. A neonate born between 37 and <42 weeks (259-293 days) of gestation.
Preterm neonate. A neonate born before 37 weeks (<259 days) of gestation irrespective of the birth weight.
124