

Ghai Essential Pediatrics8th
.pdfDiseases of Gastrointestinal System and Liver
Glycogen Storage Disorders
Glycogen storage disorders are important metabolic dis orders manifesting in childhood with varied clinical picture ranging from asymptomatic hepatomegaly (type VI) to hypoglycemia (type I, III) and decompensated end stage liver disease (type IV).
Type I glycogen storage disease (van Gierke dis ease) Inabilitytoconvertglucose-6-phosphate to glucose in the liver results in inability to mobilize glycogen. Depending on whether this is due to glucose-6-phospha tase deficiency or translocase deficiency, it is classified as type lA or lB.
Hepatomegaly, doll-like facies, hypoglycemia, seizures, growth retardation, hyperuricemia, hypertriglyceridemia andlacticacidosis aremain manifestations. Hypoglycemia is more marked after first few months of life as the fre quencyoffeedingdecreases.Liveradenomamightdevelop with risk of bleeding and malignant transformation.
Hepatomegaly and nephromegaly is appreciated on imaging.Plateletdysfunctionmaybepresent. Neutropenia is specifictotype lB. There is mildtransaminaseelevation. Liver biopsy shows hepatocytes with vacuolated cyto plasm and glycogen accumulation (PAS stain positive, diastase sensitive) along with microvesicular steatosis; fibrosisisabsent.Definitediagnosisdependsonmeasuring enzyme activity in the liver or mutational analysis.
Management hinges on providing a constant source of glucose in the form of slowly digested complex carbo hydrates. This is achieved by frequent daytime feeding, supplementation of uncooked corn starch both in day and specifically at night. As the child grows into adolescence, longer periods of fasting may be tolerated. Since the metabolism of other carbohydrates also yield glucose-6- phosphate, galactose and fructose also need to be restricted. Strict metabolic control with dietary therapy is the key to avoiding complications.
Type Ill glycogen storage disorder There is adeficiency of debranching enzyme manifesting as hepatospleno megaly, hypoglycemia, fibrosis in the liver and elevation in transaminases. Whilehypoglycemia and hepatospleno megalyimprove,80-85% develop amyopathyin typeIII a disease while the other 15% (type III b) have only liver involvement.Thesecasesaremanagedwithdietsimilarto thatintypeI GSD,exceptthathighproteindietispreferred and there is no need to restrict galactose and fructose.
Type IV glycogen storage disorder In type IV GSD there is a deficiency of branching enzyme resulting in deposition of an amylopectin like structure in the liver. The presentation is with chronic liver disease, portal hypertension andhepaticdecompensation. Most children are symptomatic by 3 yr of age. Treatment is largely supportive and liver transplantation is required for patients with advanced disease.
Suggested Reading
Clayton P. Inborn errors presenting with liver dysfunction. Semin Neonatol 2002;7:49-63
Mayatepek E, Hoffmann B, Meissner T. Inborn errors of carbo hydrate metabolism. Best Pract Res Clin Gastroenterol 2010;24:607-18
Nonalcoholic Fatty Liver Disease (NAFLD)
This is a common cause of liver disease in children and is closely associated with obesity and insulin resistance. The prevalence is increasing with the expanding prevalence of childhood obesity. NAFLD is a clinicopathological diagnosis characterized by macrovesicular steatosis in hepatocytes, in absence of other causes of chronic liver disease. It ranges from simple steatosis (macrovesicular steatosis in hepatocytes without inflammation) to non alcoholic steatohepatitis (NASH, macrovesicularsteatosis in hepatocytesassociatedwith inflammation and fibrosis) to cirrhosis of liver and hepatocellular carcinoma. Insulin resistance and hyperinsulinemia is regarded as essential to thedisease mechanism. Hyperinsulinemia is a response to energy dense diet (rich in saturated fats, sugars and refined carbohydrates). This diet elicits hyperinsulinemia, provides exogenous free fatty acids and drives the liver
towards lipogenesis. An important environmental factor is physical inactivity. Various metabolic and genetic
disorders that are associated with fatty liver disease in children are shown in Table 11.35.
Clinical presentation Most children are asymptomatic. Some have vague abdominal pain; examination shows generalized obesity, cutaneous striae and hepatomegaly. Splenomegaly is uncommon. Acanthosis nigricans, a velvety brown-to-black pigment in skin folds and axillae, typically associated with hyperinsulinemia can be found in 30-50% patients.
Investigations Typical biochemical abnormalities in NASHinclude moderatelyraisedserumaminotransferases (withALT more raised thanAST). Metabolic abnormalities include hypertriglyceridemia, elevated fasting serum insulin and hyperglycemia. Other disorders, which may cause fatty liver can be eliminated on basis of clinical and
Table 11.35: Cause of fatty liver disease in children
Metabolic |
Nonalcoholic fatty liver disease*, Alstrom |
|
syndrome, Bardet-Biedl syndrome, |
|
polycystic ovary syndrome, lipodystrophy |
|
syndromes, Prader-Willi syndrome, |
|
galactosemia*, hereditary fructose |
|
intolerance*, glycogen storage disease* |
Drugs |
Amiodarone, perhexiline, methotrexate, |
|
prednisolone, calcium channel blockers |
Others |
Craniopharyngioma, hypothalamic |
|
disorders, acute starvation, jejuno-jejunal |
|
bypass, total parenteral nutrition* |
*Common conditions

-i....E_s_s_e_n_ t_ia_i_P_e_d_ i_a _tr_ic_s_________________________________
biochemicalfindings. Thediagnosisof NAFLDissuspected when there is raised serum ALT or evidence of fatty liver on radiological studies. Liver histology is required for diagnosisof NASH. ChildrenwithNASHhaveahistologic picture of periportal inflammation, mononuclear inflam matory infiltrate, minimal ballooning of hepatocytes and few Mallory bodies.
Treatment The first step in treating NAFLD is to identify it. Besidesheightandweight,waistcircumferenceprovides highly informative data and is a surrogate for visceral obesity. The treatment has two goals: to reverse liver diseaseandto promote healthy growth. Lifestyle changes aimed at weight reduction are essential. Dietary changes andincreasedphysical activity lead to diminished insulin resistance.VitaminE,ursodeoxycholicacidandmetformin aregiventotargetinsulinresistanceandoxidantstress. The role of bariatric surgery has not been established for childhood obesity. Prevention of over-weight and obesity in children is the best strategy to overcome theproblem of
NAFLD.
Suggested Reading
Barshop NJ, Sirlin CB, Schwimmer JB, Lavine JE. Review article: epidemiology, pathogenesis and potential treatments of pediatric non alcoholic fatty liver disease. Aliment Pharrnacol Ther 2008;28:13-24
Marion AW Fatty liver disease in children Arch Dis Child 2004; 89:648-52
Drug Induced Liver Disorders
Medications are an important cause of liver dysfunction in children. A wide spectrum of liver diseases from acute liverfailuretoacute andchronichepatitisandportalhyper tensionmaybeprecipitated bydrugs. Themechanismmay be a predictable direct hepatotoxicity or more commonly an idiosyncratic drug reaction. Drug induced liver disorders may mimic allforms of liver disease. A reliable diagnosisis madeonly after detailedhistoryand exclusion of other causes of liver dysfunction. Anti-tubercular drug therapy is an important cause, followed by anti convulsants (phenytoinandcarbamazepine). Patientswith hypersensitivity features like skin rash, fever, lympha denopathy and eosinophilia have a better outcome than those without hypersensitivity. Treatment is by timely suspicion and withdrawal of the offending drug.
Suggested Reading
Devarbhavi H, Karanth D, Prasanna KS, Adarsh CK, Mallikarjun P. Drug induced liver injury with hypersensitivity features has a better outcome: a single center experience of 39 children and adolescents. Hepatology 2011;54:1344-50
Hepatic Manifestations of Systemic Diseases
Apart from disorders that directly involve the liver, a number of disorders affecting other organ systems also have hepatic manifestations. While in some disorders these manifestations may be benign, in others the hepatic manifestations might significantly affect the outcome.
lschemic Hepatitis
Severe shock may lead to hypoperfusion of the liver. This shock may be the result of sepsis, acute cardiogenic shock orsevereintraoperativehypoperfusionasincardiacsurgery. It usually manifestsasanelevationof transaminasestohigh levels. Thedegreeofhepaticinjurydependsontheduration andseverityofshock. Thus,coagulopathyas evidenced by prolonged prothrombin time and encephalopathy may result. Jaundice is a late manifestation.
Cardiac Disorders
Apart from an acute cardiogenic shock or intraoperative hypoperfusion in cardiac surgery, liver involvement may be seen as a result of congestion in right heart failure or as part of syndromesthatinvolveboth theliver and theheart.
Chronic right heart failure may lead to hepatomegaly, splenomegaly and over long periods result in cardiac cirrhosis. Alagillesyndrome, causedbysyndromicpaucity of intralobular bile ducts results in infantile cholestasis; patients also show peripheral pulmonary stenosis, tetralogy of Fallot and atrial or ventricular septa! defects. In biliary atresia, splenic malformation syndrome anomaly, there may be vascular malformations and congenital heart disease.
Sepsis and Systemic Infections
Gram positive andgram negative bacterialinfections may resultinjaundice. Uptoone-thirdofneonataljaundicehas been attributed to sepsis. The mechanism may vary from impaired canalicular bile transport due to defective transporter polarization in hepatocytes without hepatic necrosis to elevated bilirubin load due to hemolysis in clostridium infections and hepatocellular necrosis in pneumococcalinfections. Typhoidmightresultinelevated alkaline phosphatase,transaminasesandlactatedehydro genase. Transaminase elevation and liver dysfunction is also present in dengue hemorrhagic fever and malaria.
Immunological Disorders
Juvenile idiopathic arthritis may be associated with hepatomegaly and elevated transaminases. Systemic lupus erythematosus may be associated with hepato megaly or autoimmune hepatitis. Transplacental transfer of auto-antibodies might lead to neonatal SLE with transient cholestasis, congenital heart block, dermatitis and hematological abnormalities.
Hemolytic Anemias
Inthalassemia, therepeatedtransfusionsand theincreased iron absorption due to ineffective erythropoiesis leads to chronic iron overload, fibrosis and cirrhosis. Recurrent transfusionsincrease the risk of acquiring hepatitis B and hepatitis C infections. Sickle cell anemia has a similar risk oftransfusionrelatedhepatitis butmorespecificproblems are acute hepatic crisiswhich is a result ofischemic insult.

Diseases of Gastrointestinal System and Liver
Theseindividualsare also at higher risk of pigment stones resultingin acute andchroniccholecystitis.These episodes may be difficult to differentiate from acute hepatic crisis.
Malignancies
Leukemias and lymphomas might be associated with hepatic infiltration, presenting as jaundice. Hemophago cytic lymphohistiocytosis, either familial or infection induced, presents with jaundice, hepatosplenomegaly, liver dysfunction and cytopenia. It is an important differential diagnosis for liver failure in the first few monthsoflife.Sclerosing cholangitismaybe acomplication of Langerhans cell histiocytosis.
Bone Marrow Transplant
Conditioning chemotherapy and total body irradiation may lead to veno-occlusive disease manifesting as weight gain, hepatomegaly, ascites and jaundice. Other causes of liver dysfunction after bone marrow transplant include graft versus host disease (acute or chronic), sepsis, infec tions hepatitis and drug toxicity.
Endocrine Disorders
Uncontrolled diabetes mellitus presents with hepato megaly and fatty changes. Hypothyroidism manifests as jaundice in the neonatal period predominantly due to impaired conjugation of bilirubin and partly due to decreased bile flow.
Celiac Disease
Elevatedtransaminases maybe observed, which normalize with gluten free diet.
Neonatal Cholestasis
Jaundice at 2 weeks of age is a relatively common finding, seen in 2.4% to 15% newborns. While it is chiefly uncon jugated hyperbilirubinernia due to breast milk jaundice, cholestatic jaundice is an uncommon but potentially serious condition that indicateshepatobiliary dysfunction. Neonatal cholestasis is defined as direct bilirubin value greater than 1 mg/dl if thetotal bilirubin is less than 5 mg/ dl, or a value of direct bilirubin that represents more than 20% of the total bilirubin if the total bilirubin is >5 mg/dl.
The Indian Academy of Pediatrics recommends that newborn babies having jaundice beyond 14 days of age with dark colored urine with or without acholic stools should be referredtoappropriate healthfacility for further investigations and treatment without loss of time. The causes of neonatal cholestasis according to the experience of eight centers in India are shown in Table 11.36.
Clinical Features
Neonatalcholestasis is characterized by highcoloredurine along withjaundice.Asubsetofpatientspresentwithsigns of coagulopathy like skin or intracranial bleeds (seizures,
Table 11.36: Etiology of neonatal cholestasis (N=1008)
Neonatal hepatitis (47%): Idiopathic giant cell hepatitis
Infection: TORCH, sepsis malaria, urinary inlection Metabolic (4%): Galactosemia, alpha antitrypsin deficiency, TPNrelated,tyrosinemia,storage disorders,hemochromatosis
Other causes (2%): Inspissated bile plug syndrome, recurrent intrahepatic cholestasis, progressive familial intrahepatic cholestasis, hypothyroidism, Down syndrome
Obstructive (38%): Biliary atresia (34%), Choledochal cyst (4%) Ductal paucity (3%): Syndromic variety (Alagille syndrome), Nonsyndromic variety
Unknown (6%)
irritability and bulging fontanel). Hepatomegaly or hepatosplenomegaly is common. Early decompensation is afeature inpatientswithanunderlying metabolic disorder. Inasick infant, oneshouldconsiderthediagnosisofgalacto sernia, tyrosinemia, hemochromatosis, herpes and sepsis. Patients with biliary atresia and choledochal cyst are otherwise healthy looking. Bilateral cataract and E. coli sepsis is typical of galactosernia, whereas rash (maculo papular or petechial), fever, chorioretinitis, rnicrocephaly and lethargy are suggestive of congenital infections. Triangular facies, pointed chin, prominent ears, cardiac murmurs and butterfly vertebrae are seen in Alagille syn drome. Splenohepatomegaly with cherry-red spot on fundus examination suggests storage disorder.
Diagnosis
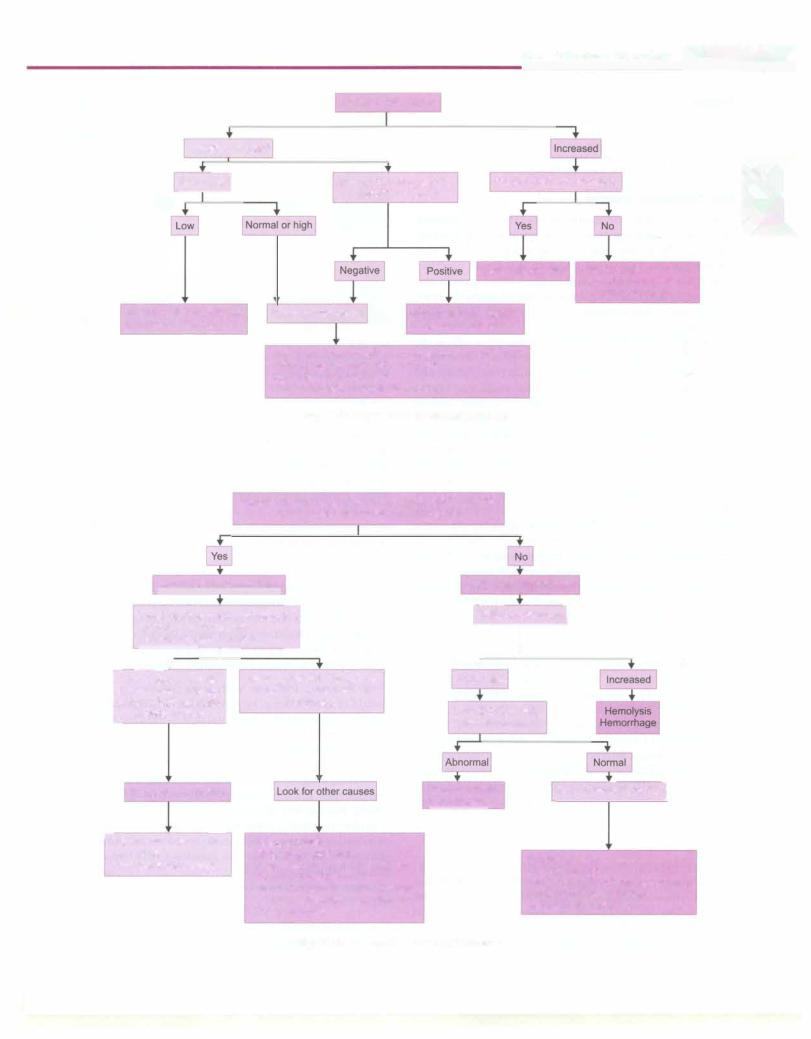
Neonatal cholestasis has multifactorial etiology (Table 11.36). Stool color is an important sign in the differential diagnosis. These infants need a detailed investigative workup based on a rational approach so as to avoid unnecessary and costly investigations. The etiology and algorithm of evaluation is differentin a 'sick' and 'not sick' infant with cholestasis as shown in the flowchart below (Fig. 11.15).
In an infant with pale stools, HIDA scan is not of discrimi natoryvalue asbothbiliaryatresiaandsevereintrahepatic cholestasis show a nonexcretory pattern, i.e. no activity in intestine at 24 hr. Priming with UDCA or phenobarbitone for 3 days before HIDA scan improves the diagnostic efficacy of HIDA. Liverbiopsy isan accurate (90-95%) test for differentiating biliary atresia from other causes of neonatal cholestasis. In biliary atresia, portaltract expan sion, ductularproliferation andfibrosisisseen, whereas in neonatalhepatitis,thereisalterationinlobulararchitecture, focal hepatocellular necrosis and giant cells formation. Laparotomy and peroperative cholangiography may be required in an infant with equivocal biopsy findings and no excretion on HIDA scan, to confirm the diagnosis of biliary atresia.
In an infant with pigmented stools, ultrasound of abdomen and liver biopsy is done to determine etiology and specific

___E_ss__eni__tal P__de__iars__------tic ----------------------------
Conjugated hyperbllirublnemla
Transaminases, albumin, y glutamyl transpeptidase. alkaline phosphatase, prothrombin time
Assess clinical- condition J Look for stool color for 3 days
|
Sick, or clinical setting |
|
|
|
|
|
|
|
|
Urine for reducing substances |
|
Abdominal ultrasonography |
|||||||
GALT assay |
|
|
|
|
|
|
|
||
Blood and urine cultures |
|
Pigmented stool |
Pale stool |
||||||
TORCH screening |
|
||||||||
cx-fetoprotein |
Normal gallbladder |
Small or nonvisualized gallbladder |
|||||||
Urine succinylacetone |
|
|
|
|
|
Triangular cord sign* |
|||
|
|
|
|
|
|||||
Ferritin, buccal mucosa! biopsy |
|
|
|
|
|
|
|
||
Malarial parasite |
|
|
|
|
|
|
|
||
Liver biopsy if indicated |
|
|
|
|
|
|
|
||
|
|
|
fConsider er biopsy |
|
|
||||
|
|
|
|
|
|
|
|
|
|
Neonatal hepatitis: Giant cell |
|
Biliary atresia on liver biopsy or liver biopsy |
|||||||
transformation, lobular disarray |
|
equivocal; no excretion on HIDA scan |
|||||||
Bile duct paucity |
|
Laparotomy and peroperative cholangiography |
|||||||
Storage disorder |
|
||||||||
Kasai portoenterostomy
Fig. 11.15: Evaluation of a patient with neonatal cholestasis. GALT galactose-1-phosphate uridyl transferase; HIDA hydroxy iminodiacetic acid *echogenic area in porta hepatitis.
treatment is done according to the cause. A detailed metabolic workup is required for infants with conditions like progressive familial intrahepatic cholestasis types I and II, tyrosinemia and bile acid synthesis defects.
Management
Delayed diagnosis leads to problems of undernutrition, coagulopathy, pruritus, portal hypertension, ascites and hepatic encephalopathy. The management is begun as soon as the child is seen, parallel to investigations.
General management This includes the following:
i.Nutritional. Adequatecaloric intake (125-150% of RDA based on ideal body weight) with medium chain tri glyceridesupplementationis necessary. Breastfeeding
should be continued and supplementation with high MCT formulae should be done; 2-3% calories should come from essential fatty acids. Nasogastric feed is offered to anorexic children. Supplementation of fat soluble and water soluble vitamins is done (Table 11.37). In addition, these infants require supplements of calcium, phosphorus and magnesium.
ii.Forinfantswithpruritus, urodeoxycholic acid (UDCA, 10-20 mg/kg/day), rifampicin (10 mg/kg/day) and cholestyramine (250 mg/kg/day) are used. UDCA is the first agent and others are used in patients with persistent symptoms.
ill. Management of other complications like ascites, gas trointestinal bleeding and hepatic encephalopathy is discussed in respective sections in the chapter.
|
Table 11.37: Multivitamin preparations for neonatal cholestasis |
|
Drug |
Dose |
Side effects |
Vitamin K |
2.5-5 mg on alternate day or 5 mg IM/IV |
None |
|
daily for 3 days and then monthly |
|
Vitamin D |
Oral 2500-5000 IU/day or 30,000 IU IM |
Hypercalcemia |
|
at diagnosis and then monthly |
Nephrocalcinosis |
Vitamin A |
Oral 2500-15,000 IU/day or 30,000 IU IM |
Hepatotoxicity, hypercalcemia, pseudotumor |
|
at diagnosis and then 10,000 IU monthly |
cerebri |
Vitamin E |
Aquasol E 50-400 IU/day |
Potentiation of vitamin K deficiency, coagulo |
|
tocopherol polyethylene glycol 1000 |
pathy, diarrhea |
|
succinate: 15-25 IU/kg/day (if available) |
|
Water soluble vitamins |
Twice the recommended daily allowance |
None |

Diseases of Gastrointestinal System and Liver
Specific management This is available only for some etiologies as follows:
i.Biliary atresia is managed by Kasai procedure (hepatoportoenterostomy). The best results are obtained if it is done early (<60 days of age) and at centers with expertise. Liver transplantation is indicated in children who fail to drain bile after Kasai procedure or have progressed to end stage cirrhosis either despite surgical treatment or due to late diagnosis.
ii.Choledochal cyst: excision of cyst and hepaticojejun- ostomy.
iii.Herpes simplex: intravenous acyclovir.
iv.Bacterial sepsis: intravenous antibiotics.
v.Toxoplasmosis: pyrimethamine andsulfadiazine with folinic acid.
vi.Galactosemia: lactose free diet.
vii.Hemochromatosis: IV immunoglobulins (IVIG) with
exchange transfusion may be useful.
There is considerable delay in referral of patients with neonatalcholestasis to higher centers inIndia. Thisresults in delayed etiologic diagnosis, missed opportunity for corrective biliary atresia surgery in first 60 days and liver decompensation in patients with metabolic etiology.
Suggested Reading
Consensus report on Neonatal cholestasis syndrome. Indian Pedi atrics 2000;37:845-51
Guideline for the evaluation of cholestatic jaundice in infants: Rec ommendations of the North American Society for Pediatric Gastroen-
terology, Hepatology and Nutrition. J Pediatr Gastroenterol Nutr 2004;39:115-28
Roberts EA. Neonatal hepatitis syndrome. Semin Neonatol 2003; 8:357-74
Liver Transplantation
Livertransplantationispossiblefor a number ofdisorders. The graft is obtained either from the cadaver or can be a split graft (leftlateralgraft or leftlobe)from aliving donor. Auxiliary liver transplantation is another method where nativeliver isnotremovedand aliver graftfromthe donor is surgically placed in addition to the native liver. This is usuallydonefor Crigler-Najjar typeI oracuteliverfailure.
Themainindicationsforlivertransplantationarebiliary atresia, fulminant hepaticfailure andchronic liver disease secondary to multiple causes and hepatic tumors. Careful selection of a blood group compatible donor is necessary, including detailed evaluation of liver functions and viral serologies. The recipient's diseased liver is removed and thenewliveristransplanted,ensuringvascularandbiliary anastomoses.Patientsrequirelifelongimmunosuppression using corticosteroids, tacrolimus and mycophenolate mofetilinitiallyandlater maintenance therapywithtacro limus. Rejection and infection are major complications following transplantation. Five-year patient survival rate exceeds 80%.
Suggested Reading
Karnath BM, Olthoff KM. Liver transplantation in children: Update 2010. Pediatr Clin N Am 2010;57:401-14

Hematological Disorders
Tulika Seth
Hematopoiesis
The stem cells from which endothelial cells and hematopoietic cells develop are called hemangioblasts. Stem cells that give rise to only blood cells, called pluripotent stem cells, divide to formtwo colony forming units (CFU), including the common myeloid precursor for granulocytes, erythrocytes, monocytes and mega karyocytes (CFU-GEMM) and the common lymphoid precursor (CFU-L) which differentiates into lymphocytes. Two cell lines arise from the CFU-GEMM, the progenitor for erythrocytes and megakaryocytes (CFU-EMk) and that for granulocytes and monocytes (CFU-GMo). Each of these further develop into specific lineages, e.g. CFU-GMo gives rise to granulocyte lineages for eosinophils (CFU-Eo), neutrophils (CFU-N), and basophils (CFU-Baso) and a lineage for monocytes (CFU-Mo). The CFU-L differentiates to form to B lymphocytes, natural killer (NK) cells, and T lymphocytes. The sequential development of hemato poietic cells is driven and regulated bylocal growthfactors and cytokines.
ANEMIA
Anemia is present when the hemoglobin level is more than two standard deviations below the mean for the child's age and sex (Tables 12.1 and 12.2). According to the third National Family Health Survey (NFHS3), 79% of Indian children have anemia, including 71% of urban children and 84% of those in rural areas.
Physiological Adaptations
Anemia results in a decrease in the oxygen-carrying capacity of blood with compensatory physiological
Table 12.2: Cutoffs for hemoglobin and hematocrit proposed by the World Health Organization to define anemia
Age group |
Hemoglobin (g/dl) |
Hematocrit % |
Children,6mo to 5yr |
<11.0 |
<33 |
Children, 5-11yr |
<11.5 |
<34 |
Children, 12-13yr |
<12.0 |
<36 |
Non-pregnant women |
<12.0 |
<36 |
Men |
<13.0 |
<39 |
Source: WHO 1997
|
Table 12.1: Hemoglobin and hematocrit in infancy and childhood |
||
|
Hemoglobin (g/dL) |
|
Hematocrit (%) |
Age |
Mean |
-2 SD |
Mean |
Birth (cord blood) |
16.5 |
13.5 |
51 |
1-3days (capillary) |
18.5 |
14.5 |
56 |
1 weeks |
17.5 |
13.5 |
54 |
2 weeks |
16.5 |
12.5 |
51 |
lmo |
14.0 |
10.0 |
43 |
2mo |
11.5 |
9.0 |
35 |
3-6mo |
11.5 |
9.5 |
35 |
0.5-2yr |
12.0 |
10.5 |
36 |
2-6yr |
12.5 |
11.5 |
37 |
6-12yr |
13.5 |
11.5 |
40 |
Girls12-18yr |
14.0 |
12.0 |
41 |
Boys 12-18yr |
14.5 |
13.0 |
43 |
Values two standard deviations below the mean (-2 SD) indicate the lower limit of normal
-2 SD
42 45 42 39 31 28 29 33 34 35 36 37
330

adjustments. When the enhanced release of oxygen from hemoglobin and increase in blood flow to the tissues is insufficient tomeet requirements, tissue hypoxia develops. Blood volume is maintained by an increase in plasma volume and redistribution of blood flow to vital organs such as brain, muscle and heart. Increase in stroke volume causesanincrease incardiac output, whichincreasesblood flow and improves oxygen delivery to tissues.
Evaluation for Etiology
The history provides significant clues to the etiology of anemia. Causes vary with age and anemia may be multifactorial. In infants, history should include that for maternal infections, anemia or collagen vascular diseases; prematurity; bloodloss; jaundice (hemolysis due to ABO or Rh incompatibility, G6PD deficiency, sepsis) and presence of hemangiomas or cephalhematoma. Infants may have physiological anemia at 2-3 months of age. In infants and young children a dietary history is useful, including type and quantity of milk and intake of hematinics. Delayed or inadequate weaning with predominantlymilk-based diet results inpoor iron intake, leading to nutritional iron deficiency, usually occurring at 6 months to 2 yr of age; other causes include chronic diarrhea or cow milk allergy. Megaloblastic anemia may be nutritional, due to use of goat milk in infants and a vegetarian diet in older children. Adolescents are particularly susceptible due to increased micronutrient requirements during the growth spurt, and in girls menstruation and/or pregnancy.
History of pica, blood loss (melena, epistaxis), drug intake (e.g. anticonvulsants), chronic diarrhea, prior surgery, chronic infections, liver or renal disease and transfusions should be taken. A family history of severe anemia and/or need for blood transfusions suggests thalassemia major. History of gallstones and recurrent jaundice is noted in various types of hemolytic anemia (e.g. hereditary spherocytosis). Anemia affecting only male members of the family points to an X-linked disorder like glucose-6-phosphate dehydrogenase (G6PD) deficiency. Lack of response toiron supplements suggests thalassemia minor.
Over 70% of patients with thalassemia major present with anemia by 6 months of age. Similarly, children with Diamond-Blackfan syndrome (pure red cell aplasia) present at about 3 months of age; diagnosis is based on persistently low reticulocyte count and absence of erythroid precursors in the bone marrow. The age at presentation for Fanconi anemia variesbetween 3-4 yr to adulthood.
Clinical Features
The hemoglobin level at which symptoms of anemia appear, depends on the rate of development of the anemia. Symptoms occur at higher hemoglobin levels if anemia develops rapidly, as with hemorrhage. The
Hematological Disorders -
most common and earliest symptoms include lassitude, and easy fatigability. Alternatively, children may have
anorexia, irritability and poor school performance. I Dyspnea on exertion, tachycardia and palpitations
indicate severe anemia. Other symptoms include dizziness, headache, tinnitus, lack of concentration and drowsiness and with severe anemia, clouding of consciousness.
The most prominent and characteristic sign is pallor, detected most reliably in nail beds, oral mucous membranes and conjunctivae. Observing palmar creases and skin is insufficient as the skin creases in children lack pigmentation.Facial pallor varies with skinpigmentation and presence of edema. A mid-systolic 'flow' murmur, chiefly in the pulmonary area, is appreciated when the degree ofanemiaincreases, reflecting increased bloodflow across heart valves. Systolic bruits and postural hypo tension may be noted with moderate to severe anemia. Severe anemia is characterized by a high output state with an elevated pulse pressure and a 'collapsing' pulse. Anemia may precipitate heart failure even with a normal cardiovascular system. Electrocardiographic changes, found in 30% patients with hemoglobin below 6 g/dl, include depressed ST segment and flattened or inverted
Twaves.
Etiological clues on examination include radial limb
abnormalities (suggest bone marrow failure syndromes), splenomegaly (hemolytic anemia, malaria, kala-azar, tuberculosis or storage diseases) and lymphadenopathy and hepatosplenomegaly (malignancies or infections). Presence of petechiae or purpura indicates concomitant thrombocytopenia, while icterus suggests liver disease or hemolysis.
Investigations
A complete hemogramis requiredtounderstandwhether the anemia is isolated or other cell lines are affected. Changes in the red cell indices provide significant information on the type of anemia and may precede loweringofhemoglobin (Table12.3). Themeancorpuscular volume (MCV) denotes the size of the red cells while the mean corpuscular hemoglobin (MCH) and mean cor puscular hemoglobin content (MCHC) provide infor mationon red cellhemoglobinization.Based on the MCV, anemia is classified as microcytic, normocytic and macrocytic (Figs 12.1 to 12.3). Patients with thalassemia minor oriron deficiencyhave lowMCV, MCH and MCHC while those with megaloblastic anemia have elevated MCV. The red cell distribution width (RDW) provides an estimate of the size differences in red blood cells. Hence, a low RDW means that the red blood cells are uniform in size, while a large RDW indicates considerable variations in the cell size.
Examination of the peripheral smear reveals red cell morphology. The presenceof schistocytes, polychromasia or parasites may help make the diagnosis. The reticulocyte

|
s -------------- |
|
__ |
E_ssentia l _Pe_diatric_-------------------- |
- |
_ |
|
|
Table 12.3: Red cell indices and serum iron studies in normal children
Red cell indices |
Birth |
0.5-2 yr |
6-12 yr |
Mean corpuscular volume |
108 |
78 |
86 |
Mean corpuscular hemoglobin |
34 |
27 |
29 |
|
|||
Mean corpuscular hemoglobin concentration |
33 |
33 |
34 |
Red cell distribution width (RDW)* |
12.8 ± 1.2% |
|
|
Serum iron |
60-170 µg/dl (10-30 µmol/1) |
||
Serum ferritin, median (range) |
100 (15-300) ng/ml (boys); 40 (15-200) |
||
Total iron binding capacity |
250-400 µg/dl (47-70 µmol/1) |
||
Transferrin saturation•• |
20-50% |
|
|
*ROW= standard deviation (SD) of red blood cell volume x 100/mean corpuscular volume. **Transferrin saturation= Serum iron x 100/total iron binding capacity
Girls, 12-18 yr |
Boys, 12-18 yr |
90 |
88 |
30 |
30 |
34 |
34 |
|
ng/ml (girls)
counthelps distinguish between anemiacaused by red cell destruction and decreased production (Table12.4). Normal reticulocyte count in newborns is 2-6% and in children 0.5-2%. However,thereticulocytecountshould becorrected for the degree of anemia. lhis is done as follows:
Corrected reticulocyte. count=Reticulocyte. count x Actual hematocrit Normal hematocrit
where the normal hematocrit is 45%. The corrected reticulocyte count should be1-2%in a healthy individual.
In patients with anemia with increased reticulocyte count, the Coomb test can help distinguish between immune and hereditary hemolytic anemia. When nutritional anemia is suspected, iron studies and levels of vitamin B12 and folic acid are determined.
Table 12.4: Reticulocyte count in evaluation of anemia Low reticulocyte count
Congenital or acquired, aplastic or hypoplastic anemia Transient eythroblastopenia of childhood
Pure red cell aplasia Parvovirus B19 infection
Bone marrow infiltration by malignancy, storage disorder
High reticulocyte count
Hemolysis
Hemorrhage Splenic sequestration
Recovery from vitamin Bl2, folic acid or iron deficiency Sepsis
Peripheral smear; reticulocyte count; iron studies
Normal or reduced reticulocyte count
Low serum iron Low ferritin Increased total iron binding capacity
Iron deficiency
Low serum iron
Normal or high ferritin
Normal or low TIBC
|
|
|
|
|
|
|
|
Increased reticulocyte count; |
|
|||||||
|
|
|
|
|
|
|
|
abnormal red cell morphology |
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
Normal or increased serum iron |
jHemoglobin ':!_PLC or electrophoresis |
||||||||||||||
|
||||||||||||||||
|
|
Normal or low total iron |
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
binding capacity and ferritin |
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
Hemoglobin |
|
|
|
|
Hemoglobinopathy |
|||||||
|
|
electrophoresis or HPLC |
|
|
|
|
hemoglobin SS, |
|||||||||
|
|
|
|
|
I |
|
|
|
|
S-C, S-13 thalassemia |
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
+ |
|
|
|
|
|
L, |
|
|||||||||
|
|
bnorma |
|
|
|
|
||||||||||
|
|
.-- --·--- |
||||||||||||||
|
|
rmal |
||||||||||||||
Hemoglobinopathies, |
Sideroblastic anemia |
|||||||||||||||
|
e.g. thalasemia |
|||||||||||||||
13 major, 13 minor, a
Erythrocyte sedimentation rate C-reactive protein
Inflammatory
disease
Fig. 12.1: Approach to microcytic anemia. HPLC high performance liquid chromatography
|
|
|
|
|
|
|
|
Hematological Disorders - |
|||||
|
|
|
|
|
|
Reticulocyte count |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Normal or reduced |
|
renal, hepatic, |
Positive tests for hemolysis |
|
||||||||
Serum |
|
Screen for |
I |
||||||||||
|
|
|
|
|
|
endocrine disease |
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|||
Anemia of chronic disease Early iron deficiency
|
Bone marrow biopsy |
Hemolytic anemia |
|
Anemia of renal, hepatic, |
|
|
|
endocrine disease |
Infiltrative disease {leukemia, metastasis, myelofibrosis) Aplastic anemia, pure red cell aplasia
Myelodysplastic syndrome Congenital dyserythropoietic anemia
Fig. 12.2: Approach to normocytic anemia
Hemorrhage
Recovery from nutritional deficiency or sepsis
Peripheral smear for macro-ovalocytes (MCV >120 fl), hypersegmented (6-lobed) neutrophils
|
|
Megaloblastic anemia likely |
|
Likely non-megaloblastic |
||||||||||||||||||
Levels of vitamin B12 and folic ac d' |
ulocyte count] |
|||||||||||||||||||||
|
(or response to therapy) |
|
|
j |
|
|
|
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||
Bone marrow examination |
|
|
|
|
|
|
|
I |
|
|
||||||||||||
|
|
|
|
-,- |
|
No megaloblastic changes in |
|
|
|
|
|
|
|
|||||||||
__+ |
|
|
|
|
|
|
|
|||||||||||||||
|
|
|
|
|
+ |
|
|
|
|
|
|
|
||||||||||
Low levels or anemia |
|
|
|
Decreased |
|
|
|
|
|
|
||||||||||||
[mproves with therapy; |
|
|
|
|
marrow or no improvement |
|
|
|
|
|
|
|
|
|
|
|
||||||
megaloblastic changes |
|
|
|
|
|
|
with therapy |
Liver function tests, |
||||||||||||||
in bone marrow |
|
|
|
|
|
|
|
thyroid hormone |
||||||||||||||
|
|
|
|
|
||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Megalobastic anemia |
|
|
|
|
Hypothyroidism |
|
Bone marro aspiration] |
|
||||||||||||||
|
|
|
|
Liver disease |
|
|
||||||||||||||||
F"Therapy with vitamin B12 |
|
|
Hypothyroidism |
|
|
|
|
|
|
|
|
|
|
|
||||||||
and folate |
|
|
|
|
|
|
|
Drugs (e.g., phenytoin, |
|
|
|
|
|
Aplastic anemia |
||||||||
Treat underlying cause |
|
6 mercaptopurine, sulphonamides) |
|
|
|
|
|
|||||||||||||||
|
|
|
|
|
|
|
|
|
|
Congenital deficiency of |
|
|
|
|
|
Red cell aplasia |
||||||
|
|
|
|
|
|
|
|
|
|
transcobalamin II or intrinsic factor |
|
|
|
|
|
Congenital dyserythropoietic anemia |
||||||
|
|
|
|
|
|
|
|
|
|
Cyanotic congenital heart disease |
|
|
|
|
|
Myelodysplastic syndrome |
||||||
|
|
|
|
|
|
|
|
|
|
Down syndrome |
|
|
|
|
|
Sideroblastlc anemia |
||||||
Fig. 12.3: Approach to macrocytic anemia

.....E_s_s_e_n_ ti a_i_P_e_d_ i a _tric_s_________________________________
-
Sugg ested Reading
National Family Health Survey III, Government of India, 2007; http://www.nfhsindia.org/abt.html, accessed March 2012
Orkin SH, Nathan DG, Ginsburg D, Look AT, Fisher DE, Lux SE. (Eds.) (2009). Nathan and Oski's Hematology ofInfancy and Childhood (7th Ed.). Philadelphia, PA: Saunders Elsevier
Iron Deficien cy An emia
Iron is essential for multiple metabolic processes, including oxygen transport, DNA synthesis and electron transport. In severe iron deficiency, low levels of iron containing enzymes affect immune and tissue function. Iron deficiency can also result in diminished growth and learning. Irondeficiencyanemia occurs when the decrease in total iron body content is severe enough to diminish erythropoiesis and cause anemia.
Body iron is regulated carefully by absorptive cells in the proximalsmall intestine,whichalterironabsorptiontomatch body losses of iron. Iron deficiency results from diminished dietary iron absorption in the proximal small intestine or excessivelossesofbodyiron.Irondeficiencyinolderchildren is usually caused by dietary deficiency; the absorption of iron is further impaired by dietary constituents that lower the absorption of non-heme iron, e.g. phytates, phosphates and tannates. Intercurrent infections such as hookworm infestation and malaria worsen the problem. Healthy newborns havebodyiron storesof 250 mg or approximately 80 parts per million (ppm); this decreases to approximately 60 ppm in the first 6 months of life, as milk is a poor source of iron. Infants consuming cow milk are more likely to have iron deficiency than breastfed infants because (i) the iron contained in cow milk has lower bioavailability; (ii) bovine milk has a higher concentration of calcium that competes with iron for absorption; and (iii) due to gastrointestinal blood loss with cow milk allergy.
Evaluation
A careful dietary history is important, including the type of milk and weaning foods in infants and the use of supplements. Pica increases the risks of lead poisoning and helminthic infections.
Clinical findings are related to the severity and rate of development of anemia. Irritability and anorexia usually precedeweakness,fatigue, leg cramps,breathlessnessand tachycardia.Congestivecardiac failure and splenomegaly may occur with severe untreated anemia. Angular stomatitis, glossitis, koilonychia and platynychiaarenoted in severe cases.
Investigations
The peripheral blood smear reveals microcytic, hypochromic red cells (Fig. 12.4) with anisocytosis and poikilocytosis andan increased redcell distribution width (RDW). The MCV and MCHC are reduced. The total number of red cells is reduced, unlike in thalassemia where it is increased. The serum level of iron and ferritin
Fig. 12.4: Peripheral smear from a child with iron deficiency anemia, shows microcytosis (the red blood cells are smaller than the small lymphocyte in the field), hypochromia (central pallor >1/3rd of cell diameter), thrombocytosis, and a few ovalocytes and tear drop cells (moderate anisopoikilocytosis). Jenner-Giemsa x 1000
are reduced while the total iron binding capacity is increased (Table 12.3). The saturation of transferrin is reduced to less than 16%. The reduction in serum ferritin occurs early and correlates well with the total body iron stores. Rarely, ferritin may be falsely normal or elevated in a sick child since it is an acute phase reactant that increases in inflammatory conditions. High free erythro protoporphyrin precedes the development of anemia.
Treatment
The cause of anemia should be identified and corrected. Hookworminfestationis the mostcommon cause ofoccult gastrointestinal bloodlossin rural India at all ages. Dietary counseling and treatmentof causative factorsare required to prevent recurrence or failure of therapy. Close follow up is required to assess for adequate response.
Oral therapy Patients with iron deficiency anemia should receive 3-6mg/kg/day ofelemental iron. Ferrous sulfate, containing 20% elemental iron,isthe most effective and economical oral preparation. Absorption is best when taken on an empty stomach or in between meals. About 10-20% patients develop gastrointestinal side effects such as nausea, epigastric discomfort, vomiting, constipation anddiarrhea. Enteric-coatedpreparations have fewer side effects, but are less efficacious and more expensive. The reticulocyte count is expected to increase within 72-96 hr of initiating therapy. After correction of anemia, oral iron should be continued for 4-6 months to replenish iron stores. Patients that show an inadequate response to therapy should be evaluated for conditions listed in Table 12.5.
Parenteral therapy The indications of parenteral iron therapy are: (i) intolerance to oral iron, (ii) malabsorption,