

Ghai Essential Pediatrics8th
.pdfStreptococcus pneumoniae). They are also at risk of other
common infectious organisms such as Salmonella,
Mycoplasma pneun10niae, Staphylococcus aureus and Escherichia coli.
Laboratory Studies
Anemia and thrombocytosis are commonly found. Leukocytosis occurs in patients with sickle cell anemia. However, a rise in the white blood cell count (i.e. >20,000 per mm3) with a left shift is indicative of infection. In the peripheral smear, sickle-shaped red blood cells are found along with target cells. Presence of Howell-Jolly bodies indicates that the patient is functionally asplenic. The baseline indirect bilirubin level may be elevated because of chronic hemolysis.
If the diagnosis of sickle cell anemia has not been made, a sickling test will establish the presence of sickle hemoglobin. Hemoglobin electrophoresis is the test that candifferentiatebetween individuals whoarehomozygous or heterozygous. Hemoglobin in a homozygous patient will chiefly (80-90%) be hemoglobin SS (HbSS), while carriers will have 35-40% as HbSS. This needs to be checked prior to blood transfusions.
Assessment During Acute Illness
In a sick child, a type and cross-match is required for probable transfusion. A chest X-ray and X-rays of bones may be indicated in pain crisis. Blood culture should be sent. Monitoring of oxygen saturation and arterial blood gases should be ordered in patients with respiratory distress. A major drop in hemoglobin (more than 2 g/dl) from baseline indicates a splenic sequestration or aplastic crisis. Reticulocyte count and examination of spleen size will help to differentiate between these two conditions. An electrocardiogram must be performed if symptoms of chest pain and/or pulse irregularities are noted.
Inpatient Management
Hydration and analgesia are the mainstays of treatment in a pain crisis. Narcotic analgesia is most frequently used. Hydration is corrected orallyif the patient is not vomiting and can tolerate oral fluids. In severe dehydration, intravenous fluids are required. Care is taken not to overload the patient and accurate intake-output monitoring should be ensured. Blood transfusion isuseful in patients in aplastic crisis and acute sequestration crisis.
Oxygen supplementation is of benefit if the patient has hypoxia. Intubation and mechanical ventilation may be required in children in whom cerebrovascular accidents have occurred, or with acute chest syndrome. Exchange blood transfusions are indicated incases of cerebrovascular accidentsand acutechestsyndrome. Thisinvolves replacing the patient's red blood cells by normal donor red blood cells, decreasing HbSS to less than 30%. They may be performed in patients with acute sequestration crisis or in
Hematological Disorders -
priapism that does not resolve after adequate hydration and analgesia.
All children require prophylaxis with penicillin or I amoxicillin, at least until 5 yr of age and should receive
Preventive Care
immunizations with pneumococcal, meningococcal and Haemophilus influenzae B vaccines. They should receive life
long folate supplementation. Hydroxyurea is a cytotoxic agent which can increaseHbF and reduce episodesofpain crisesandacutechestsyndrome and may be useful beyond 5 yr of age. Parents need to learn how to identify compli- cations and be informed for necessity and indications for admission. Patients need to be screened regularly for development of gallstones. Geneticcounseling and testing should be offered to the family.
Suggested Reading
Sachdeva A, Sharma SC, Yadav SP. Sickle cell disease. In: IAP Specialty series on Pediatric Hematology & Oncology 2006;77-96
Steinberg MH. Management of sickle cell disease. N Engl J Med 1999;40:1021-30
Aplastic Anemia
Aplastic anemia comprises a group of disorders of the hematopoietic stem cells resulting in the suppression of one or more of erythroid, myeloid and megakaryocytic cell lines. The condition may be inherited or acquired. In developed countries, bone marrow failure due to hypoplastic or aplastic anemia affects 2-6 individuals per million populations. Although precise information is lacking, the prevalence is estimated to be higher in India.
Etiopathogenesis
Hematopoietic stem cells may be deficient due to (i) an acquired injury from viruses, toxins or chemicals; (ii) abnormal marrow microenvironment; (iii) immuno logic suppression of hematopoiesis (mediated by antibodiesorcytotoxic T cells); and (iv) mutationsingenes controlling hematopoiesis resulting in inherited bone marrow failure syndromes (Table 12.12).
Clinical Features
Physical examination in case of severe anemia reveals pallor and/or signs of congestive heart failure. Ecchymoses, petechiae, gum bleeding and nose bleeds are associated with thrombocytopenia. Fever, pneumonia or sepsis may occur due to neutropenia. Inherited bone mar row failure syndromes, usually diagnosed in childhood or as young adults, may be associated with characteristic congenital physical anomalies, positive family history or neonatal thrombocytopenia. The child should be evaluated for the stigmata of congenital bone marrow failure syndromes (Table 12.12; Figs 12.9 and 12.10). However, Fanconi anemia may be present even without any abnormal phenotypic features. History of exposure

E s s |
|
s |
|
|
|
___________ |
__ |
||
___ |
|
en t iai P ed iat ric |
|
______________ |
|
|
_ |
||
|
_ _ |
_ _ _ _ _ _ |
_ _ _ _ _ _ _______ |
|
|
|
|
|
|
|
|
|
Table 12.12: Congenital syndromes associated with bone marrow failure |
|
|
||||
Syndrome |
|
|
Inheritance |
|
Associated features |
Risk of malignancy |
|
|
|
Associated with pancytopenia |
|
|
|
|
|
|
|||
Fanconi anemia |
|
AR |
|
Absent thumbs,absent radius, |
High risk of acute myeloid |
|
|
||
|
|
|
|
|
|
microcephaly, renal anomalies, |
leukemia, myelodysplasia, oral or |
|
|
|
|
|
|
|
|
short stature, cafe au lait spots, |
liver cancer |
|
|
|
|
|
|
|
|
skin pigmentation |
|
|
|
Dyskeratosis congenita |
|
X-linked recessive, |
Dystrophic nails, leukoplakia |
Skin cancer (usually squamous cell), |
|
||||
|
|
|
|
AD,AR |
|
|
myelodysplasia |
|
|
Single lineage cytopenias
Amegakaryocytic AR thrombocytopenia
Diamond-Blackfan syndrome AD,AR (pure red cell aplasia)
Thrombocytopenia absent radii AR
AR autosomal recessive; AD autosomal dominant
None |
None |
Short stature, congenital |
Leukemia, myelodysplasia, other |
anomalies in one-third, |
cancers |
macrocytosis,elevated fetal |
|
hemoglobin, raised adenosine |
|
deaminase |
|
Absent radius |
None |
to toxins, drugs like chloramphenicol, environmental hazards and viral infections, e.g. hepatitis B or C, suggest an acquired aplasia.
Laboratory Studies
Hematological features of bone marrow failure include pancytopenia or bilineage involvement, noted in aplastic anemia, single cytopenia as seen in pure red cell aplasia and amegakaryocytic thrombocytopenic purpura. Single lineage cytopenias should be differentiated from transient erythroblastopenia of childhood. Peripheral blood smear reveals anemia, occasionally with macrocytosis (<llO fl), thrombocytopenia and agranulocytosis. The corrected reticulocyte count is less than 1%, indicating reduced red cell production. Bone marrow aspirate and biopsy are
essential for evaluation of bone marrow cellularity (Fig. 12.ll). Usually,themarrow contains veryfewhema topoietic cells and is replaced with fat cells and lympho cytes.
Specific tests The sucrose hemolysis test or Ham's test may be positive in patient with underlying paroxysmal nocturnal hemoglobinuria (PNH), in which red cells are lysed by patient's acidified sera, and in congenital dyserythropoeitic anemia type II, where red cells are lysed by other acidified sera but not patient's sera. Recent transfusion with packed red blood cells may give false negativeresults. A more specific test for PNH is the assay for two complement regulatory proteins present on red blood cells, CD55 (decay accelerating factor, DAF) and CD59 (membrane inhibitor of reactive lysis, MIRL). The
Fig. 12.9: Child with Fanconi anemia. The child had hyper pigmentation, microcephaly and microphthalmia. She also had radial ray defects and growth retardation
Fig. 12.10: Radial ray defects present in a wide spectrum and include absent or hypoplastic thumbs. Thenar hypoplasia may be missed unless carefully examined

Pancytopenia
|
|
|
|
|
|
|
Peripheral blood smear, reticulocyte count, I |
||||
|
red cell indices, indirect bilirubin |
||||
+ |
|
i- |
.. |
|
|
Reduced reticulocyte
count, normal indirect bilirubin, no evidence
of hemolysis
i
Deficiency of vitamin B12 or folate Bone marrow failure or fibrosis Bone marrow infiltration (infection, myelodysplasia) Storage disorders, e.g., Gaucher disease
Hemolysis or |
Blasts on |
l |
1- |
reticulocytosis |
peripheral |
|
smear |
Sepsis |
Leukemia |
Hypersplenism |
Lymphoma |
Paroxysmal |
|
nocturnal |
|
hemoglobinuria |
|
Fig 12.11: Algorithm for evaluation of pancytopenia
peripheralbloodcellsinFanconianemiashowacharacteristic hypersensitivity towards chromosomal breakage when incubated with DNA cross-linking agents such as mitomycin C. Chromosomal fragility is noted even in patients who lack the characteristic physical stigmata of Fanconi anemia.
Treatment
Supportive care should be instituted with packed red cells for severe anemia, platelets transfusions for severe thrombocytopenia and antibiotics for management of infections. Hematopoietic stem cell transplant (HSCT) is the only definitive therapy. Criteria for referral for HSCT include: (i) young age; (ii) severe aplastic anemia; and (iii) availability of matched sibling. Patients with severe acquired aplastic anemia who cannot undergo HSCT may benefit from infusions of antithymocyte globulin (ATC) or antilymphocyte globulin (ALG) along with oral cyclosporine. Patients with neutropenia with infection should receive a trial of granulocyte colony stimulating factor (G-CSF). However, G-CSF should be discontinued after seven days even if neutrophil count does not improve, since its prolonged use carries the risk of malignant transformation.
Immunosuppressive therapy is contraindicated in children with Fanconi anemia. In these patients, hematopoietic stem cell transplantation is the only cure. However, these children continue to be at risk of malignancies inthefuture. Oralandrogenshave beenused as palliative therapy in such patients.
Prognosis
The severity and extent of cytopenias determine prognosis. Patients withsevereaplasiaare atrisk ofhigh outputcardiac failure due to anemia, bacterial andfungal infections due to neutropenia and severe bleeding due to thrombocytopenia. With current transplantation regimens, longterm survival
Hematological Disorders --
in patients with severe aplastic anemia is 60-70%, with survival rates exceeding 80% in favorable subgroups.
Suggested Reading
Varma N, Varma S, Marwaha RK, et al. Multiple constitutional aetiological factors in bone marrow failure syndrome (BMFS) in patients from north India. Indian J Med Res 2006:124:51--6
HEMATOPOIETIC STEM CELL TRANSPLANTATION-------
Bone marrow transplantation, more appropriately termed hematopoietic stem cell transplantation (HSCT), is life saving forseveralmalignantandnon-malignantdisorders. The term 'autologous' transplant is used when stem cells are harvested from the patient and 'allogeneic' when stem cells are collected from a donor, either a human leukocyte antigen(HLA)-matchedsibling or an unrelated person. The commonly used sources of hematopoietic stem cells are cytokine-mobilized peripheral blood, bone marrow and umbilical cord blood.
Indications
The indications for hematopoietic stem cell transplan tationarelistedinTable12.13. In malignant disorders, the transplant serves to rescue the bone marrow from the myelotoxic effects of the high doses of chemotherapy or radiation used to cure the malignancy. In allogeneic HSCT, the immunological response of the 'graft versus cancer effect' contributes to controlling the disease. In non-malignant diseases, the abnormal marrow is destro yedandreplacedbythehealthyunaffecteddonor marrow that corrects genetic or acquired diseases of blood cells.
Allogeneic HSCT
Donor Requirement
The ideal donor for an allogeneic transplant is a HLA identical sibling. Despite HLA identity, variations in the minor histocompatibility loci cause antigenic differences that may cause graft rejection or graft versushost disease. While transplants using partially HLA-matched siblings or an unrelated HLA-identical donors may be performed, the complications including graft versus host disease and
Table 12.13: Indications for stem cell transplantation
Malignant disorders |
Nonmalignant disorders |
Acute myeloid leukemia |
Thalassemia |
Chronic myeloid leukemia |
Aplastic anemia |
Acute lymphoblashc leukemia |
Fanconi anemia |
(high risk) |
Immunodeficiency |
Hodgkin disease |
syndromes |
Non-Hodgkin lymphoma |
Inborn errors of metabolism |
(relapsed or refractory) |
Autoimmune diseases (rare) |
Neuroblastoma |
|
Ewing sarcoma |
|
Myelodysplashc syndromes |
|
Gliomas |
|
Other solid tumors |
|

__Ess entia_l_Pe_diat_rics__________________________________ _
graft rejection are usually very severe. Most centers in Indiaconductonlyrelatedtransplants. Unlikeotherorgan transplants, ABO blood group compatibility is not essential for HSCT. Successful hematopoietic trans plantation causes the blood group of the recipient to change to that of the donor.
Conditioning Procedure
Mye/oablative conditioning In preparation for HSCT, high doses of chemotherapy are administered to suppress the bone marrow in order to (i) eradicate malignant cells or abnormal clones; (ii) suppress the host immune responses to avoid allograft rejection; and (ii) clear a 'physical space' to allow adequate growth of donor stem cells.
Non-mye/oablative conditioning Alternatively, knowing that the curative potential of allogeneic HSCT is mediated in part by an immune-mediated graft-versus tumor effect, donor T cells are used to eradicate both non malignantandmalignantcells of hostorigin without using myeloablative conditioning regimens. This form of conditioning suppresses the host's immunity sufficiently to allow allogenic engraftment without destroying the host marrow, and with lower toxicity.
Technica/ Aspects
The donor marrow is harvested under general or spinal anesthesia by repeated aspiration from the posterior iliac crests. The minimum number of marrow cells required for successfulengraftmentis estimated to be 1-3 x 108 cells per kilogram of recipient's body weight. When transfused through peripheral veins, the donor marrow cells home into the host marrow space and start engrafting. Engraftrnentisconsidered successful when the peripheral absolute neutrophil count exceeds 500/mm3 on three successive days.
After transplantation of the marrow, it takes about 2-3 weeks before engraftment occurs. During this period
intensive support and protective isolation are required. Infections, predominantly bacterial and fungal infections, remain an important cause of morbidity and mortality in patients undergoing HSCT.
Until engraftment occurs,patientsrequiremultiple red cell and platelet transfusions during the 2-4 week period of pancytopenia. Patients are profoundly immune-sup pressed and at risk of developing transfusion associated graft versus host disease (GVHD) after receiving cellular blood products. All cellular blood products should be irradiated prior to transfusion to inactivate the donor lymphocytes.
Graft Versus Host Disease
This unique complication may occur in allogeneic bone marrow transplant recipients in one two forms, acute and chronic.
Acute graft versus host disease (GVHD) This occurs within the first 3 months after transplant. It classically affects three tissues, namely the skin, gut and liver, and may be accompanied by fever. The severity can be graded according to the extent of skin involvement, degree of hyperbilirubinemia and severity of diarrhea.
Chronic GVHD This develops later than 100 days after transplant and often follows acute GVHD, but may also develop de nova. It is classified as limited or extensive chronic GVHD. Clinically, it resembles autoimmune disorders (like scleroderma) withskinrash, sicca complex, sclerosing bronchioloitis and hepatic dysfunction. The mortality varies from 20-40%. Management is with immunosuppressive agents.
Autologous Stem Cell Transplantation
Transplantation using autologous bone marrow or peripheral blood stem cells is performed similar to allogeneic HSCT, but using the patient's own stem cells for engraftment. The patient's marrow or stem cells are collected prior to chemotherapy and are subsequently used to 'rescue' the patient from the myelosuppression following the chemotherapy. The procedure is useful only for malignancies which are sensitive to chemotherapy or radiotherapy, e.g.leukemias, lymphomas, neuroblastoma and other solid tumors. Virtually all autologous stem cell transplantations are conducted using peripheral blood stem cell transplantations instead of the bone marrow since the engraftrnent is more rapid. The advantage of autologous transplantation over allogeneic transplants is the absence of GVHD and lower likelihood of graft rejection once engraftment occurs.
Peripheral Blood Stem Cell Transplantion
The procedure for peripheral blood stem cell trans plantation issimilar to bone marrow transplant except for differencesinthemethodof collectionofthestemcells and slight changes in the engraftment potential. Such transplantscanbeautologousorallogeneic. Theperipheral blood contains a small proportion (about 0.1%) of stem cells, which can be increased by administration of colony stimulating factors such as G-CSF. For allogeneic donors, G-CSF isadministeredfor4to5days, whichresultsin high numbers ofcirculatingstemcellswhichcanbecollectedby a cell separator (apheresis) machine over 2-4 hours using a large-bore venous access. This process avoids hospital admission, anesthesia and pain associated with marrow aspiration. Autologous transplantation requires the stem cellstobe collected in a similar fashion, but chemotherapy is required prior to the harvest to reduce tumor contamination andtoyieldahighproportionofstemcells.
Cord Blood Stem Cell Transplantation
Bloodfromtheplacentalcord,usuallydiscardedin clinical
practice, is a useful source for allogeneic hematopoietic

stem cells. The mainlimitation of cord blood is the limited number of nucleated cells per unit, these being 1 log less than in a bone marrow transplant. As compared to bone marrow transplantation cord blood transplantation is associated with prolonged time to engraftment with the duration to neutrophilic engraftment being about 30 and 50 days, respectively. This, along with higher incidence of non-engraftment, leads to high transplant-related mortality. The main advantage of cord blood trans plantationis alowerincidence and severity of graft versus hostdisease, which allows for 1 to 2 HLAantigenmismatch even in unrelated transplantation.
Suggested Reading
Copelan EA. Hematopoietic stem cell transplantation. N Engl J Med 2006;354:1813-26
DISORDERS OF HEMOSTASIS AND THROMBOSIS Approach to a Bleeding Child
Bleedingmayoccurdue to defects in platelets (Tables12.14 and 12.15), coagulation disorders (Table 12.16) or dysfunctional fibrinolysis. Clinical assessment, of type of bleeding, history of antecedent events and screening tests can help identify the cause, so that specific management can be initiated.
Pathogenesis
The process of hemostasis involves platelets, vessel wall and plasma proteins in a fine balance between blood flow and local responses to vascular injury (clotting). The plasma proteins involved in hemostasis perform three processes: (i) a multiple-step zymogen pathway leading to thrombin generation; (ii) thrombin-induced formation of fibrin clot; and (iii) complex fibrinolytic mechanisms that limit clot propagation. The result of these processes is the generation of insoluble fibrin and activation of platelets to form a hemostatic plug. This process is
Table 12.14: Causes of thrombocytopenia
Idiopathic thrombocytopenic purpura
Infections: Disseminated intravascular coagulation, malaria, kala-azar, dengue hemorrhagic fever, hepatitis B and C, HIV, congenital (TORCH) infections, infection associated hemophagocytosis syndrome
Medications: Valproate, penicillins, heparin, quinine, digoxin
Thrombotic microangiopatlzy: Thrombotic thrombocytopenic purpura; hemolytic uremic syndrome
Malignancies: Leukemia, lymphoma, neuroblastoma
Autoimmune or related disorders: Systemic lupus erythematoses, Evans syndrome, antiphospholipid syndrome, neonatal immune thrombocytopenia
Immunodeficiency: Wiskott Aldrich syndrome, HIV/AIDS Bone marrow failure: Thrombocytopenia with absent radii, Fanconi anemia, Shwachman-Diamond syndrome Marrow replacement: Osteopetrosis, Gaucher disease
Others: Hypersplenism, Kasabach Merritt syndrome
Hematological Disorders -
Table 12.15: Qualitative disorders of platelet function Inherited disorders
Glanzmann thrombasthenia (GP Ib deficiency) Bernard Soulier syndrome (GP 1Ib-Illa deficiency) Gray platelet syndrome
Dense body deficiency
Acquired disorders
Medications Chronic renal failure
Cardiopulmonary bypass
Table 12.16: Common coagulation disorders
Inherited disorders
Hemophilia A and B von Willebrand disease
Specific factor deficiencies Factor VII, X, XIll deficiency Afibrinogenemia
Acquired disorders
Liver disease
Vitamin K deficiency
Warfarin overdose
Disseminated intravascular coagulation
controlled by multiple proand anticoagulant pathways, platelet number and function, vascular factors and other metabolic processes. The coagulation cascade is often depictedasinvolving two pathways, intrinsicandextrinsic (Fig. 12.12). The extrinsic pathway is the primaryinitiating pathway for coagulation and is measured by prothrombin time, while the intrinsic system works as a regulatory amplification loop, measured by activated partial thromboplastin time.
Clinical Evaluation
The age ofonset ofbleeding,type ofbleeding,precipitating factors (spontaneous or following dental extraction, surgery or circumcision)assist in definingthecause (Table 12.17). In case of recent onset bleeding, history of antecedent infections, rash (Henoch-Schonlein purpura, varicella), icterus (liver failure); and prodromal diarrhea or associated renal failure (hemolytic uremic syndrome) are important. Medications associated with bleeding include anticonvulsants, penicillins, warfarin, aspirin, non-steroidal anti-inflammatory medications andheparin. History of blood transfusions helps in assessing the severity of the illness. Family history is important. The sex of affected family members and details of bleeding manifestations should be noted; a disorder affecting only boys suggests an X-linked disorder such as hemophilia, while girls may have bleeding disorders in autosomal dominant conditions like von Willebrand disease. Poor wound healing andprolongedbleeding from theumbilical stump suggests factor XIII deficiency.

|
E |
s s |
en t |
ia |
P ed iat rics |
|
_______________________ |
|||
|
__ |
_ _ _ |
_ _ |
_ _ |
_ _ _ _ _ _ _ ____________ |
|||||
|
||||||||||
|
Intrinsic Pathway |
|
|
|
Extrinsic Pathway |
management. The activated partial thromboplastin time |
||||
|
Contact activation |
|
|
|
|
|
|
deficiencies; the degree of deficiency dictates the |
||
|
|
|
|
|
|
VII |
its ratio to a contrast with an international normalized |
|||
|
Xll--x1+Xlla-.x1a |
|
|
|
|
|
standard (INR) are used to assess therapeutic warfarin |
|||
|
|
|
|
|
|
|
|
|
|
is usedto monitor heparin therapy. Prothrombin time and |
+ |
|
|
|
|
|
|
|
1 ;, ; ;;;to, |
in its reproducibility and reliability. This test is abnormal |
|
|
1x-.1xa |
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
+----VIia |
effect. Bleeding time is now rarely used due to problems |
|
|
|
+ Calcium |
|
X |
in presence of thrombocytopenia {Table 12.14) as well as |
|||||
|
|
|
l |
Ehlers Danlos syndrome {Table 12.18). Sick patients |
||||||
|
|
|
|
|
|
|
in vascular abnormalities, e.g. vasculitis in Henoch |
|||
+ |
|
Xa |
|
|
require evaluation for disseminated intravascular |
|||||
Platelet factor |
|
|
||||||||
|
|
|
|
VIiia |
1 |
l:ium + |
|
Schonlein purpura and connective tissue defects like |
||
|
|
|
|
|
|
- |
Bleeding time has been largely replaced by platelet |
|||
|
|
|
|
|
|
|
|
Platelet factor |
aggregation studies for inherited and acquired platelet |
|
|
|
|
|
|
|
|
|
|
Factor XIII |
dysfunctions. The tests required to evaluate for von |
|
[Prothrombin (factor II)----+[Thrombin (Ila) ---._1 |
coagulopathy. |
||||||||
|
Willebrand disease are von Willebrand cofactor assay, |
|||||||||
|
|
|
Fibrinogen (factor I) ------• Fibrin |
quantitation ofvon Willebrand antigen, factor VIII assay, |
||||||
|
|
|
|
|
|
|
|
1 |
Ila |
ABO blood group and electrophoretic analysis of von |
Willebrand multimers.
Fibrin clot
Fig. 12.12: In vivo coagulation cascade
Examination is done for the presence of ecchymoses (Fig. 12.13),petechia,vascularmalformations (hemangioma, telangiectasia) and rashes. The presence of splenomegaly suggests the presence of infections, malignancy, collagen vasculardisordersor hypersplenismrather than a primary bleeding defect. Rashes may be seen following drug exposure, due to infections, collagen vascular disorders, Langerhans cell histiocytosis and Wiskott-Aldrich syndrome.
Laboratory Investigations
A complete hemogram is done for platelet count, mor phology of platelets and red cells and evidence of rnicro angiopathic hemolysis (Fig. 12.14). Initial screening tests are prothrombin and activated partial thromboplastin time. Specific assays can be done to identify factor
Fig. 12.13: Large ecchymotic patch on the upper limb ofa young girl with von Willebrand disease
Table 12.17: Differences in bleeding patterns between platelet disorders and coagulation disorders
|
Platelet disorders |
Coagulation disorder |
Site of bleeding |
Skin, mucous membranes (mucosal bleeds: |
Soft tissues, joints, muscles (deep bleeds) |
|
epistaxis, oral, gastrointestinal tract) |
|
Petechiae |
Yes |
No |
Ecchymoses |
Small, superficial |
Large, deep |
Hemarthrosis, muscle bleeding |
Extremely rare |
Common |
Bleeding after minor trauma |
Yes |
No |
Bleeding after surgery |
Immediate; usually mild |
Delayed (1-2 days); often severe |
Example |
von Willebrand disease, idiopathic |
Hemophilia |
|
thrombocytopenic purpura |
|

Hematological Disorders -
History and physical examination
Platelet count
Thrombocytopenia
[Consid
Infection
Hypersplenism
Immune thrombocytopenia Thrombotic microangiopathy Malignancy
Fanconi anemia Thrombocytopenia with absent radii
Platelet dysfunction
Normal platelet count
Prothrombin time (PT), activated partial thromboplastin time (aPTT)
Oral anticoagulants Liver dysfunction Vitamin K deficiency
Disseminated intravascular coagulation
I |
+ |
|
|
normalPT |
|
Abnormal aPn1 |
||
|
Normal PT J |
I N rmal aPTT |
Hemophilia A and B |
t |
|
Anticoagulant therapy |
||
von Willebrand |
Factor VII deficiency |
|
disease |
|
|
Fig. 12.14: Work up in a child with bleeding
Table 12.18: Vascular causes of bleeding
Henoch Schonlein purpura
Vasculitis in systemic lupus erythematosus
Ehler Danlos syndrome
Scurvy
Prolonged steroid use; Cushing disease
Hereditary hemorrhagic telangiectasia
Suggested Reading
Lusher JM. Clinical and laboratory approach to the patient with bleeding. Nathan and Oski's Hematology of Infancy and Childhood, 6th edn. Eds Nathan DG, Orkin SH, Ginsburg D, Look AT. Saunders, Philadelphia, 2003;1515-26
Idiopathic Thrombocytopenic Purpura
Idiopathic thrombocytopenic purpura (ITP), renamed as immune thrombocytopenia based on evidence of auto antibody mediated consumption of platelets, is the commonest bleeding disorder presenting in children. The illness usually presents between 1 and 7 yr of age. Thrombocytopenia is termed acute if lasting less than 6 months; more than 6 months is considered chronic. It is important to correctly diagnose this entity and differen tiate it from seriousconditions like leukemia. The majority of children (60-75%) are likely to have acute ITP that resolves within 2--4 months of diagnosis, regardless of therapy.
Pathogenesis
ITP is proposed to be secondary to antibodies directed againsttheplateletglycoprotein IIb/Illacomplex. Platelets with antibodies on their surface are trapped in the spleen,
where they are removed by splenic macrophages. The mechanism of production of the antibodies is not known. Recent data describes a Thl dominant pro-inflammatory cytokine state in these individuals. Increased megakary ocyte number in the bone marrow is the hallmark of immune-mediated platelet destruction. However, a relative decrease in megakaryocyte production due to specific anti-platelet autoantibodies is also described.
Clinical Evaluation
There is often an antecedent history of febrile illness, but the patient is usually afebrile at presentation. There is a seasonal clustering of cases, the illness being more frequent duringchangeofseasons. The childpresents with sudden appearance of bruises and mucosal bleeding, epistaxis, oral oozing and prolonged bleeds with superficial trauma. The durationofsymptoms isrecorded to distinguish acute from chronic ITP.
Physical examination reveals the presence of petechiae and ecchymoses. Symptoms and signs depend on the platelet count. Bleeding is usually mild unless the platelet count drops below 20,000/µl. With platelet counts of 20,000-50,000/µl, petechiaeandecchymoses are observed following mildtrauma. There are no dysmorphic features, bony anomalies or hyperpigmentation. The presence of lymphadenopathy or splenomegaly suggests secondary causes of thrombocytopenia rather than ITP.
Laboratory Evaluation
The complete blood count shows low platelet count; other hematologicalparametersarenormal. A peripheralsmear is done to examine for abnormal cells (such as blasts) or
|
|
n i |
i i |
|
___________ |
|
|
|
s |
|
|
|
Ess e tai Pe_datr c_ |
________ |
_ |
||
|
__ |
|
______________ |
|
|
|
|
|
|
||
malarial parasites to estimate the platelet count and to exclude spurious thrombocytopenia. Circulatoryplatelets are larger in size, indicating increased production. Liver and renal function tests and lactate dehydrogenase levels aredonetoruleouthepatitis,occultmalignancy,hemolysis and hemolytic uremic syndrome. Appropriate evaluation for infection isnecessary in febrile patients. Screening tests for disseminated intravascular coagulopathy are done if sepsis is suspected. Bone marrow examination shows increased megakaryocytes and excludes marrow infiltration, leukemia or bone marrow failure.
Management
Platelet transfusions should be avoided. Minimizing the risk of hemorrhage and decreasing the long term side effects of treatment are the goals of therapy. A patient with a few scattered petechiae or bruises, platelet count above 20,000/mm3 and no mucosal bleeding only requires close observation. For children with active bleeding, treatment includes intravenous immunoglobulin 1 g/kg/day for 1-2 days, or 50-75 mg/kg of anti-0 immunoglobulin in Rh positive children. Corticosteroids can be administered after the possibility of hematological malignancyhas been ruled out on bone marrow examination. Prednisone is given at 1-4 mg/kg/day for 2-4 weeks and then tapered. Dexamethasone is administered at 20 mg/m2 over 4 days every 3 weeks for 4-6 courses. If serious hemorrhage occurs, platelet transfusions may be used under cover of steroids. Therapeutic options for chronic ITP include prednisone administered at low dose on alternate days and, in refractory cases, combinations of the following options danazol, vincristine, cyclosporine, azathioprine, rituximab (anti-CD20 monoclonal antibody), splenectomy and, most recently, thrombopoietin receptor-binding agents.
Suggested Reading
Nugent D. ASHEducation Book 2006.www.asheducationbook.org/ cgi/reprint/2006/1/97/pdf
Sharma SK, Gupta N, Seth T, et al. Successful management of re fractory chronic immune thrombocytopenia with intracranial hemor rhage by emergency splenectomy. Indian J Pediatr 2012;79:397-8
Neonatal Allo immuneThrombocytopenia
In neonatal alloimmune thrombocytopenia, fetal platelets are destroyed by maternal antibodies against paternally inheritedantigenspresent on fetalplatelets. Antibodies are usually directed against platelet antigens HPA-la or HPA-Sb. Almost half the cases occur with the first pregnancy, without history of sensitization.
While patients with mild thrombocytopenia remain asymptomatic, hemorrhagic complications, including intracranial hemorrhage, may occur within hours of birth. A high index of suspicion is required to correctly identify this condition. As specific tests are limited, the condition is primarily diagnosed by exclusion of other etiologies of
thrombocytopenia. In a sick newborn, these include sepsis, meconium aspiration and intrauterine infections; in a well looking infant, one must exclude the effect of maternal medications or maternallupus erythematosus.
Postnatal management requires transfusion of washed maternal platelets (preferably irradiated) and close monitoring until the platelet counts normalize. The risk for neonatal alloimmune thrombocytopenia increases in subsequent pregnancies. The fetus requires serial ultrasound examinations for intracranial hemorrhage. Administration of intravenous immunoglobulin (1 g/kg repeatedantenatally everyfourweeks)duringpregnancy and at birth, along with oral dexamethasone may be useful.
Hemophilia
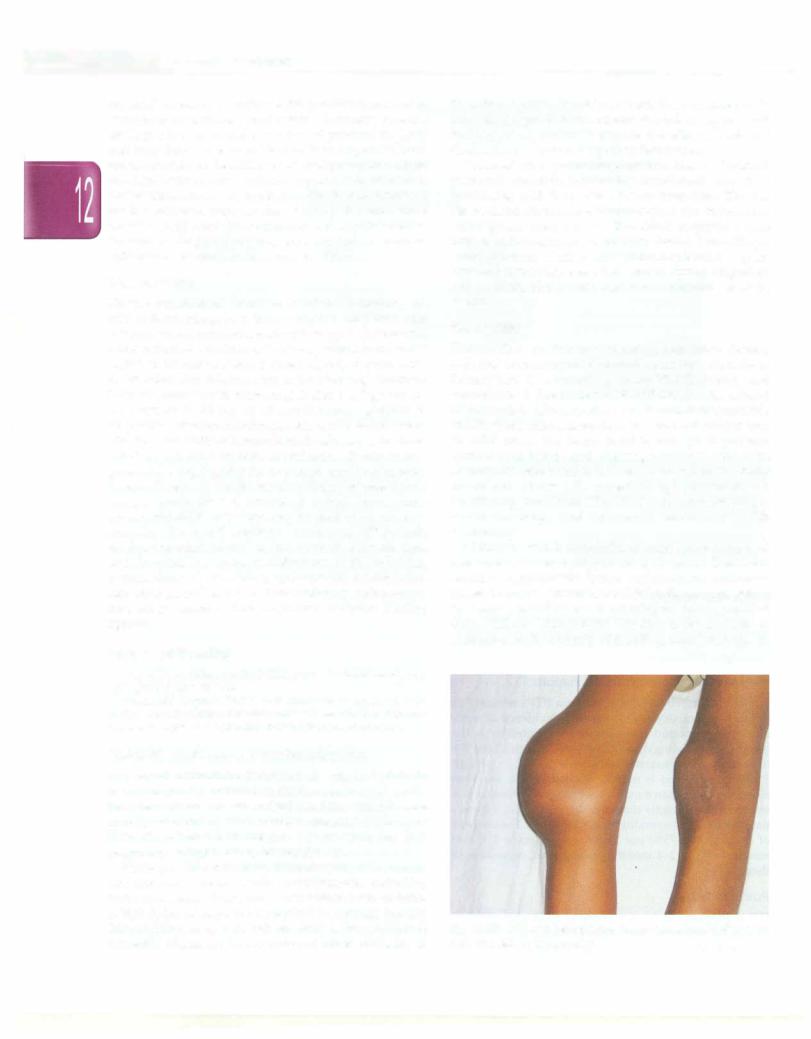
Hemophilias are the most common hereditary clotting defects, occurring as X-linked recessive disorders. Hemophilia A is caused by factor VIII deficiency and hemophilia B due to factor IX deficiency. The clinical manifestationsofhemophiliaA andBare indistinguishable and the presentation dependson the level of factor present. In mild cases, the factor level is enough to prevent spontaneous bleeds and bleeding manifests only with surgery orseveretrauma.Inseverecases,wherethe factor levels are below 1%, repeated, spontaneous and debilitating joint bleeds (Fig. 12.15) may occur, leading to severe handicaps, and intracranial bleeds may be life threatening.
Children with hemophilia should be managed at specialized centers equipped for their needs. Treatment requires appropriate factor replacement, judicious physiotherapy toprevent chronicjoint disease, counseling for injury prevention and monitoring for development of factor VIII and IX inhibitors. The dose of factor replaced is targeted to the severity of bleeding manifestations. In
Fig. 12.15: Child with hemophilia with knee hemarthrosis with severe pain and signs of inflammation

major bleeds, e.g. intracranial hemorrhage, the target for factor level is correction to 80-100% of normal value, for hemarthroses, the target is 30-50% of normal values. One unit of factor VIII per kg body weight increases the level of factor VIII by 2%. The principles of therapy of hemophilia B are similar; except that factor IX is used for replacement, one unit of factor IX per kg raises factor level by only 0.7%. Therefore the doses of factors required are calculated as follows:
Dose of factor VIII=% desired rise in FVIII x body weight (kg) x 0.5 Dose of factor IX=% desired rise in FIX x body weight (kg) x 1.4
Hence, a patient with severe hemophilia A and hemarthroses requires 15 U/kg of factor VIII every 12-24 hours for 1-2 days, while the dose needed in a patient with intracranial bleeding is 40-50 U/kg every 12 hours for approximately 7-14 days. However, lower doses may be effective. E-aminocaproic acid or tranexamic acid may be effective as adjunct therapy in mild cases of hemophilia.
Replacement therapy for children with hemophilia with concentrates of factor VIII or IX is expensive and often not available. Cryoprecipitate and fresh frozen plasma (FFP) can be used to control bleeding but carry the risk of transmitting HIV and hepatitis B and C. Cryoprecipitate contains factor VIII, fibrinogen and von Willebrand factor but not factor IX, and is not useful in patients with hemophilia B. When fresh plasma is frozen, it retains all factors at hemostatic levels, including labile factors V and VII.
Patients with severe hemophilia (lessthan 1% measurable factor level), may be given factor replacement two to three times a week to reduce the risk of bleeds. This results in less deformity and allows the child to play normally. While this is an expensive mode of treatment, it provides good quality of life. All children should receive hepatitis B immunization; vaccines can be given by the subcutaneous route and the parents should be counseled regarding injury prevention. Genetic counseling is required and families should be informed of the availability of prenatal diagnosis.
Suggested Reading
Roberts HR, Key N, Escobar MA. Hemophilia A and hemophilia B.
In: Williams Hematology 8th eds Ed. Kaushansky K, Litchman MA, Beutler E, et al. McGraw-Hill Companies, New York, 2010;2009-29
Vitamin K Deficiency
Phytomenadione or vitamin K1 plays a vital role in the production of vitamin K dependent coagulation factors, including factors II (prothrombin), VII, IX and X, and proteins C and S. Vitamin K is found in green leafy vegetables and oils (soyabean, canola) and is synthesized by colonic bacteria. Deficiency is frequent in newborns due to poor transmission of vitamin K across the placenta, its paucity in breast milk, lack of gut bacteria and prematurity of liver function. Later in life, vitamin K deficiency may follow prolongedantibiotic use, parenchymal liver disease,
Hematological Disorders -
prolonged total parenteral nutrition and malabsorption. Bleeding due to classic vitamin K deficiency occurs in 0.25- 1.7% of infants. The prevalence of late vitamin K deficiency bleeding in breastfed infants not given prophylaxis is 20 cases per 100,000 live births.
Deficiency of vitamin K dependent factors leads to prolonged prothrombin and activated partial thromboplastin time. Precise diagnosis, by measuring proteins induced by vitamin K absence (PIVKA), is usually not required. Vitamin K is administered as a single subcutaneous dose of 1 mg at birth to prevent hemorrhagic disease of the newborn. Prophylaxis with vitamin K is widely practiced and safe. Larger doses of vitamin K (2- 10 mg) can be given to treat symptomatic neonates who did not receive prophylaxis or have anticoagulant overdose; this is repeated till coagulation studies are normal. Fresh frozen plasma is administered if there is overt bleeding, or liver dysfunction is suspected.
Suggested Reading
American Academy of Pediatrics Committee on Fetus and New
born. Controversies concerning vitamin K and the newborn. Pediat rics. 2003; 112: 191-2
Disseminated lntravascular Coagulopathy
Disseminated intravascular coagulation (DIC) is an acquired dysregulation of hemostasis. The presentation ranges from an isolated derangement of laboratory parameters to severe bleeding from multiple sites, associated with high mortality. DIC may be triggered by a variety of conditions that result in activation of the clotting cascade, deposition of fibrin in the micro circulation and consumption of platelets and clotting factors. The diagnosis of DIC is clinical (Fig. 12.16); laboratory tests provide confirmatory evidence.
Pathophysiology
There are three main pathologic processes involved.
Fig. 12.16: An ill child with disseminated intravascular coagulation. Shows ecchymoses, purpura and subconjunctival hemorrhage

|
s --------- |
|
__ |
E_ssent ail _Pe_diatric_------------------------ |
- |
_ |
|
|
Thrombin generation in DIC is mediated by the extrinsic (tissue factor) pathway. The tissue factor accumulates on activated platelets by binding to platelet P-selectin which results in thrombin generation.
Amplification role of thrombin Thrombin generated amplifies inflammation and clotting by activating platelets and factors V, VIII and IX, which lead to more thrombin production. Activated factor XIII leads to it cross-linking with fibrin clots making them insoluble, while thrombin activates the fibrinolysis inhibitor, making the clot resistant to fibrinolysis.
Propagation of fibrin deposition There is suppression of fibrinolysis secondary to sustained increase in plasma levels of plasminogen-activator inhibitor. Following injury, infection or other precipitating factors, there is releaseofcytokines (tumornecrosis factor alpha, IL-1 and IL-6)whichchangethe endothelium from an anticoagulant to a procoagulant surface and interfere with fibrinolysis.
As DIC continues, fibrinogen, prothrombin, platelets and other clotting factors are consumed beyond the capacity of the body to compensate and bleeding ensues. Activated protein C has an anti-inflammatory effect; it downregulates the tissue factor and decreases calcium ion flux.
Causes
The main illnesses causing disseminated intravascular coagulopathy are listed in Table 12.19. Acute DIC is usually associated with severe infections. Chronic DIC occurs due to an intermittent activation of DIC by giant hemangiomas,certainvasculitis and in some solidtumors.
Clinical and Laboratory Evaluation
DIC is characterized predominantly by bleeding manifestations in critically ill patients.
Screening tests Peripheral blood film examination and hemogram show schistocytes and thrombocytopenia. Prothrombin time, activated partial thromboplastin time and thrombin time are prolonged.
Supportive tests Increase in fibrin degradation products or D-dimersischaracteristic. No singletestisdiagnostic of DIC. The presence of thrombocytopenia and low levels of fibrinogen (50% drop in either value) are most sensitive in making a laboratory diagnosis. A DIC scoring system has been established based on the recommendations of the Scientific Standardization Committee of the International Society on Thrombosis and Hemostasis (Table 12.20); An underlyingpredisposing disorder is a prerequisite for the use of this algorithm. A score of ;::,5 is diagnostic.
Treatment
The underlying disease must be managed appropriately. In cases of sepsis, antibiotics are necessary. In snake bites,
Table 12.19: Disorders which cause disseminated intravascular coagulopathy (DIC)
Acute DIC |
Chronic DIC |
Medical conditions |
|
Septicemia or infections* |
Solid tumors |
Fulminant hepatic failure |
Kasabach-Merritt |
Heat stroke, hyperpyrexia |
syndrome |
Severe burns |
Liver cirrhosis |
Acute promyelocytic |
|
leukemia, neuroblastoma |
|
Snake bite |
|
Collagen vascular disorders |
|
Surgical conditions |
|
Severe trauma-crush injury, |
Vascular tumors |
multiple fractures with fat |
Aortic aneurysm |
emboli |
|
Major operations |
|
Severe renal allograft rejection |
|
Iatrogenic |
|
Hemolytic transfusion |
Artificial surfaces |
reaction; massive transfusion |
|
Heparin induced thrombosis |
|
* Includes the following infections:
Bacterial: Meningococcus, gram-negative bacteria, group B streptococus
Viral: Arboviruses, varicella, variola, rubella, paramyxo- viruses, HIV, Ebola virus
Parasitic: Malaria
Mycotic: Candida, aspergillus
Rickettsial: Rocky Mountain spotted fever
anti-snake venom should be administered. Tissue per fusion and respiratory function must be maintained by replacement with intravenous fluid and provision of oxy gentocorrecthypoxia.Coagulopathymaybecompounded by vitamin K deficiency, which requires correction.
Hemostatic support (replacement therapy) In patients who have low levels of platelets, fibrinogen and otherclotting factors as revealed by deranged coagulation tests, replacement of deficient components is useful. Replacement therapy isnotindicated if there is no clinical bleeding and if no invasive procedures are planned. Monitoring is essential for guiding management and checking adequacy of replacement component support. The different bloodcomponentsavailable and commonly used in DIC are: fresh frozen plasma, cryoprecipitate, platelet concentrates and packed red cells (Table 12.21). The required doses depend on rate and degree of consumption. Replacement therapy can be halted when stabilization in platelet counts and fibrinogen levels and a fall in fibrin degradation products is observed.
Heparin therapy Heparin therapy has not proved to be useful and may be harmful in most cases of acute DIC.