

Кислоты и основания
В современной органической химии широко используются две теории кислот и оснований: теория Бренстеда и теория Льюиса.
Теория Бренстеда
Согласно теории Бренстеда, кислота является донором, а основание акцептором протона. Для взаимодействия с протоном основание должно иметь пару электронов (это может быть неподеленная электронная пара, -орбиталь или даже доступная по энергии -орбиталь). Когда кислота отдает протон, остающаяся частица сохраняет электронную пару и, по крайней мере, теоретически, является основанием. Его называют сопряженным основанием данной кислоты. С другой стороны любое основание, присоединившее протон, потенциально способно его отдать, следовательно, протонированное основание является кислотой. Всем кислотам соответствуют сопряженные основания, а всем основаниям – сопряженные кислоты. Любую кислотно-основную реакцию можно описать уравнением:
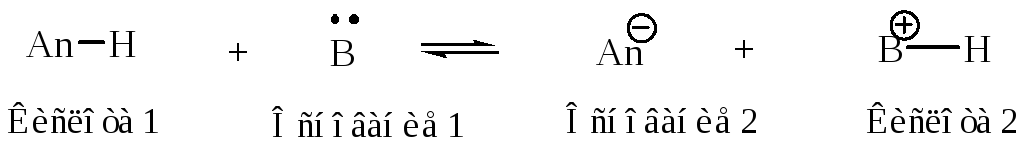
Заряд кислоты всегда на +1 превышает заряд сопряженного основания. Многие вещества могут одновременно обладать свойствами основания и кислоты; такие соединения называются амфотерными.
Силу кислот можно определить как меру способности отдавать протон, а силу основания – как меру способности присоединять протон. Кислотно-основные реакции проходят благодаря тому, что сила разных кислот неодинакова. Если, например, такую кислоту, как НСl ввести в контакт с сопряженным основанием более слабой кислоты, произойдет обмен протоном, т. к. HCl более склонна отдавать протон, нежели, например, CН3CО2Н.

Вообще равновесие кислотно-основной реакции всегда сдвинуто в сторону образования более слабой кислоты и более слабого основания. Такая реакция всегда протекает в случае двух кислот, поэтому, определяя положение равновесия, можно измерить относительную силу кислот и соответственно оснований. Все кислоты и основания можно расположить в определенном порядке по мере уменьшения их кислотности или основности. Точную силу кислот и оснований, выраженную в рКа, можно измерить только для кислот более слабых, чем Н3О+, и более сильных, чем Н2О. Для остальных кислот рКа измерить очень трудно, а кислотность в таких случаях оценивается приблизительно по косвенным данным. Необходимо отметить, что сила кислот в значительной мере зависит от растворителя. Например, HCl в воде более сильная кислота, чем пикриновая, а в ДМФА – наоборот. Порядок кислотности может сильно изменяться при переходе в газовую фазу. Например, в газовой фазе толуол более сильная кислота, чем Н2О.
Влияние строения молекул на их кислотность и основность
Строение молекулы может оказывать самое различное влияние на ее кислотность или основность. Обычно в большинстве молекул одновременно действуют два или более разных эффектов, различить которые обычно очень трудно или невозможно.
Полярный эффект. В качестве примера влияния полярного эффекта на кислотность можно сравнить рКа уксусной и нитроуксусной кислот:

Различие в строении этих молекул в том, что водород метильной группы уксусной кислоты замещен на группу NO2 в нитроуксусной кислоте. Будучи сильным акцептором электронов, NO2-группа оттягивает электронную плотность от отрицательно заряженной группы СО2- в анионе нитроуксусной кислоты, тем самым стабилизирует анион, в результате чего эта кислота оказывается почти в 1000 раз сильнее уксусной кислоты. Вообще любой эффект, выражающийся в оттягивании электронной плотности от отрицательно заряженного центра, является стабилизирующим по отношению к аниону, поскольку он размазывает заряд и, следовательно, повышает кислотность. Таким образом, можно утверждать, что группы, оттягивающие электроны за счет полярного эффекта, повышают кислотность и понижают основность, тогда как электронодонорные группы действуют в противоположном направлении.
Резонансные эффекты. Резонанс, который стабилизирует основание, но не его сопряженную кислоту, повышает кислотность, и наоборот. Примером служит более высокая кислотность карбоновых кислот по сравнению со спиртами:

Ион RСО2- стабилизирован резонансом в отличие от иона RСН2О-. Такой же эффект наблюдается и в других соединениях, содержащих группы С=О или С=N. Так, кислотность амидов выше, чем аминов, кислотность сложных эфиров выше, чем простых эфиров, а кислотность кетонов выше, чем алканов. Резонансные эффекты играют важную роль.
Корреляция с периодической таблицей. При сравнении кислот и оснований Бренстеда, отличающихся положением элемента в периодической таблице, наблюдаются следующие закономерности:
а) при движении вдоль ряда слева направо кислотность повышается, а основность понижается. Таким образом, кислотность повышается в ряду CH4 < NH3 < H2O < HF, а основность уменьшается в ряду CH3- > NH2- > OH- > F-. Такая закономерность обусловлена изменением электроотрицательности. Этот же фактор обуславливает большую разность кислотности в ряду RCOOH >> RCONH2 >> RCOCH3;
б) кислотность повышается, основность понижается при движении вниз в группах, несмотря на то, что электроотрицательность уменьшается. Эта закономерность связана с увеличением размеров атома. Например, кислотность повышается в ряду HF < HCl < HBr < HI, а основность повышается в ряду NH3 > PH3 > AsH3. Это правило справедливо для Н2О и других протонных полярных растворителей. В других растворителях порядок может быть иным;
в) те кислоты, которым требуется только одна электронная пара для завершения октета, более сильны, чем те, которым требуется две электронные пары. Так, например GaCl3 более сильная кислота, чем ZnCl2. Это правило применимо для кислот Льюиса.
Статистические эффекты. В случае симметричных двухосновных кислот константа первичной диссоциации вдвое больше, поскольку имеется два эквивалентных водорода, способных к ионизации. Константа вторичной диссоциации только наполовину больше ожидаемой. Отношение К1/ К2 4.
Водородная связь. На силу кислот и оснований большое влияние оказывает внутримолекулярная водородная связь. Например, разница кислотности орто- и пара-гидроксибензойных кислот:

Такая разница в кислотности объясняется тем, что внутримолекулярная водородная связь между группами ОН и СО2- орто-изомера стабилизирует анион, а в пара-изомере возможна только межмолекулярная водородная связь, которая стабилизирует анион в меньшей степени. В общем случае стабилизирующее действие водородной связи в сопряженных основаниях повышает кислотность соответствующих кислот.
Пространственные эффекты. Стерические эффекты могут влиять на кислотность или основность не прямо, а посредством резонанса. Например, орто-трет-бутилбензойная кислота почти в 10 раз сильнее пара-изомера благодаря тому, что орто-заместитель выталкивает группу СО2- из сопряжения с бензольным кольцом. Благодаря этому эффекту орто-бензойные кислоты сильнее соответствующих пара-бензойных кислот независимо от того, являются заместители электроноакцепторными или электронодонорными группами.
Тип гибридизации атома. Поскольку энергия s-орбитали ниже энергии р-орбитали, то, чем больший s-характер будет иметь гибридная орбиталь, тем ниже будет ее энергия и в большей степени будет стабилизирован карбанион при сопряженном основании. Следовательно, сильнее будут соответствующие кислоты. Устойчивость карбанионов падает в ряду HCC- > H2C=CH- > H3C–CH2-, а кислотность уменьшается в ряду HCCH > H2C=CH2> H3C–CH3.
Влияние среды на силу кислот и оснований
Среда (растворитель) оказывает очень значительное влияние на силу кислот и оснований. Эффект растворителя заключается в различной сольватации молекул растворенного вещества. Например, если основание сольватируется в большей степени, чем его сопряженная кислота, его устойчивость по сравнению с сопряженной кислотой повышается, и основность такие растворители снижают.
Так, метиламин по отношению к Н+ более сильное основание, чем аммиак, а диметиламин – еще более сильное основание. Это можно объяснить электронодонорным эффектом группы СН3. Однако триметиламин, который в таком случае должен был бы быть еще более сильным основанием, в действительности более слабое основание, чем диметил- и метиламин. Это явление можно объяснить различной гидратацией. Ион NH4+ благодаря наличию положительного заряда намного лучше гидратирован, чем молекула NH3. В результате этого эффекта сила основания повышается на 11 единиц рКа. При переходе от водорода к метильным группам это различие в гидратации становится меньше и разница в силе основания для триметиламина только 6 единиц рКа. Таким образом, имеет место действие двух эффектов в противоположном направлении: за счет эффекта поля основность повышается при увеличении числа метильных групп, а за счет эффекта гидратации она уменьшается. Если два этих эффекта суммируются, например, в воде, самым сильным основанием оказывается Me2NH, а самым слабым NH3. В газовой фазе, где эффекты сольватации отсутствуют, основность аминов убывает в ряду Me3N > Me2NH > MeNH2 > NH3 в соответствии только с действием полярного эффекта.