

37_DFU_2.1 / DFU_Dopolnenie_2cr
.pdfКориці цейлонської кори олія
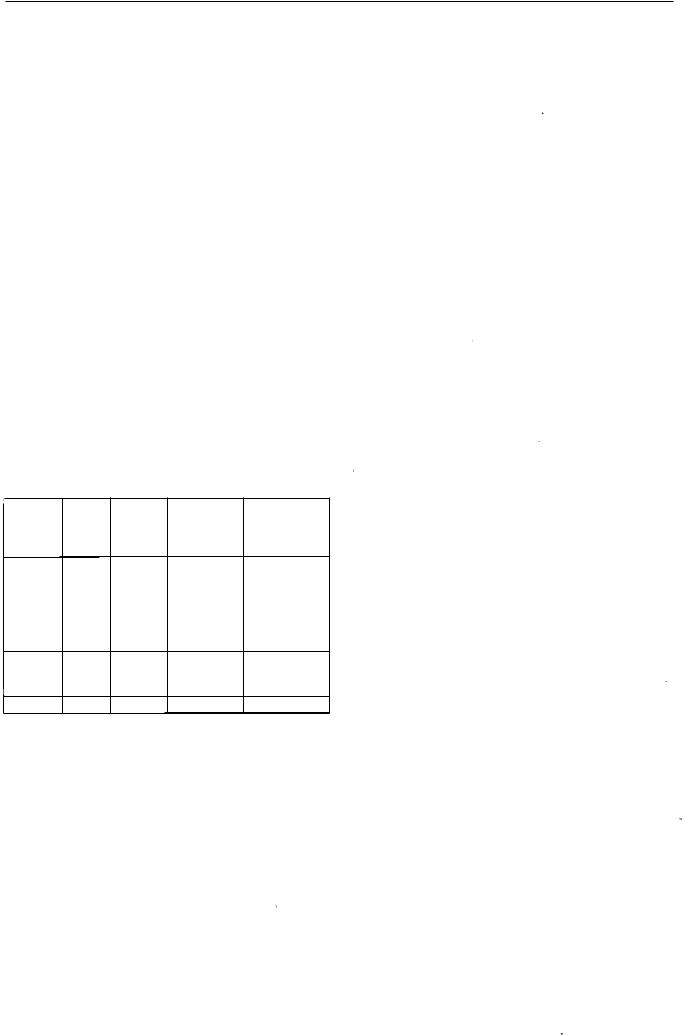
Використовуютьтаку програмутемпературногорежи му:
|
|
Темпера- |
|
Швидкість |
|
||
|
Час |
підвищення |
|
||||
|
|
||||||
|
тура |
Примітки |
|||||
|
(хв) |
температури |
|||||
|
ес) |
|
|||||
|
|
|
ес/хв) |
|
|||
|
|
|
|
|
|
||
Колонка |
0 - 1 0 |
60 |
|
- |
|
ізотерміч- |
|
|
|
|
|
|
|
|
ний режим |
|
1 0 - 75 |
60 1 90 |
2 |
|
лінійний |
||
|
75 - 1 60 |
|
|
|
|
|
градієнт |
|
190 |
|
|
|
|
ізотерміч- |
|
|
|
|
|
|
|
|
ний режим |
|
|
|
|
|
|
|
|
Блок вводу |
|
200 |
|
|
|
|
|
проб |
|
|
|
|
|
|
|
Детектор |
|
240 |
|
|
|
|
|
|
|
|
|
|
|
|
|
Хроматографують 0.2 мкл розчину порівняння. При хроматографуванні зазазначенихумов порядок вихо ду піків має відповідати порядку зазначення речовин ускладі розчину порівняння. Залежно відумов прове дення випробуваl-fНЯта стану колонки, пік кумарину може виходити передабопісля піка транс-2-метокси коричногоальдегіду. Відмічаютьчасиутримування цих субстанцій.
Хроматографічна система вважається придатною, якщо коефіцієнт розділення для піків кумарину і транс-2-метоксикоричного альдегіду становить не менше 1 .5.
Хроматографують 0.2 мкл випробовуваного розчину. Використовуючи часи утримування, визначені із хро матограми розчину порівняння, визначаютьположен ня компонентів розчину порівняння на хроматограмі випробовуваного розчину.
Визначають вміст кожного компонента, у відсотках, методом внутрішньої нормалізації.
Вміст компонентів, увідсотках, маєзнаходитисяута кихмежах:
-транс-коричний альдегід: від 70 % до 90 %,
-цинамілацетат: від 1 .0 % до 6.0 %,
-евгенол: менше 0.5 %,
-кумарин: від 1 .5 % до 4.0 %,
-транс-2-метоксикоричний альдегід: від 3.0 % до 15 %.
ЗБЕРІГАННЯ
У повітронепроникному максимально наповненому контейнері, у захищеному від світла та нагрівання місці.
КОРИЦІ ЦЕЙЛОНСЬКОї КОРИ ОЛІЯ
Сіппатоті zeylanicii corticis aetheroleum
CINNAMONBARKOIL, CEYWN
Ефірна олія, одержана із кори гілок Сіnnатотит zey/anicum Nees ( с . Verum J.S. Presl.) методом перегон
ки з водяною парою.
ВЛАСТИВОСТІ
Опис. Прозора, рухома рідина світло-жовтого кольо ру, що протягом часу стає червонуватою, із характер ним запахом коричного альдегіду.
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: В. Друга ідентифікація:А.
А. Визначення проводять методом тонкошаровоїхро матографії (2.2.27), використовуючи ТШХпластинки із шаром сuлікагелю Р.
Випробовуваний розчин. І мл субстанції розчиняють в ацетоні Рідоводятьоб'єм розчинутим самим розчин никомдо 10 мл.
Розчин порівняння. 50 мкл транс-коричного альдегіду Р, І О мкл евгенолу Р, ІО мклліналолу Рі І О мкл /З-каріофі лену Ррозчиняютьу 96 %спирті Рі доводять об'єм роз чину тим самим розчинником до 10 мл.
Налінію стартухроматографічної пластинки смугами наносять 10 мкл випробовуваного розчину та 10 мкл розчину порівняння. Пластинку поміщають у камеру із сумішшю розчинниківметанол Р - толуол Р( І0:90). Коли фронт розчинників пройде 15 см відлініїстарту, пластинку виймають із камери, сушать на повітрі, об прискуютьрозчином анісового альдегіду Р і перегляда ють при денному світлі при нагрівання при темпера турі від 1 ОО а с до І05 ас протягом 5- 1О хв.
Нахроматограмі випробовуваного розчинумають виявлятися зони на рівні зон на хроматограмі розчину  порівняння, відповідніїм за забарвленням.
порівняння, відповідніїм за забарвленням.
В. Переглядають хроматограму, одержану у випробу ванні нахроматографічний профіль. Часи утримуван ня основних піків на хроматограмі випробовуваного розчину мають співпадати із часами утримування ос новних піків на хроматограмі розчину порівняння. Ліки, відповідні сафролу, кумаринута цинеолу, можуть бути відсутніми нахроматограмі випробовуваногороз чину.
472 |
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 1 .2 |

Кофеїн
лену Ррозчиняютьу 96 %сnиртіРідоводять об'єм роз чину тим самим розчинником до 1О мл.
Пластинка: ТШХпластинка із шаром сuлікагелю Р.
Рухома фаза: метанол Р - толуол Р ( 10:90).
Об'єм проби, що наноситься: 10 мкл, смугами.
Відстань, що має пройти рухома фаза: 15 см від лінії старту.
Висушування: на повітрі.
Виявлення: обприскуютьрозчином анісового альдегіду Р
і переглядають при денному світл.ї при нагріванні при температурі від 1ОО ос до 105 ос протягом 5-1 О хв.
Результати: на хроматограмі випробовуваного розчи нумаютьвиявлятися зони нарівні зон нахроматограмі розчину порівняння, відповідні їм за забарвленням. Зона, відповідна транс-коричному альдегіду, може бути дужеслабко забарвленою або відсутньою.
В. Переглядають хроматограму, одержану у випробу ванні на хроматографічний профіль.
Результати: часи утримування характерних піків на хроматограмі випробовуваногорозчину маютьспівпа дати ізчасами утримування характерних піків на хро маТ0грамі розчину порівняння. Піки, що відповідають цинеолу, сафролу, транс-коричному альдегіду, цина мілацетатутакумарину, можутьбути відсутніми нахро матограмі випробовуваного розчину.
ВИПРОБУВАННЯ НА Ч ИСТОТУ
Відносна густина (2.2.5). Від 1 .030 до 1 .059.
Показник заломлення (2.2.6). Від 1 .527 до 1 .540.
Оптичне обертання (2.2. 7). Від -2.50 до +2.00.
Хроматографічний профіль. Газова хроматографія
(2.2.28): метод внутрішньої нормалізації.
Випробовуванийрозчин. Випробовувана субстанція.
Розчин порівняння. І О мкл цинеолу Р, 1О мкл ліналолу Р, 10 мкл {З-каріофілену Р, 10 мкл сафролуР, 10 мкл транс коричного альдегіду Р, І О мкл цинамілацетату р, І ОО мкл
евгенолу Р, 10 мг кумарину Ррозчиняють в 1 мл ацето-
ну Р.
Колонка:
-матеріал: кварц,
-розмір: 60 м Х 0.25 мм,
-нерухома фаза: макрогол 20000Р.
Газ-носій: гелій для хроматографіїР.
Лінійна швидкість газу-носія: 1 .5 мл/хв. Поділ потоку: І : І ОО.
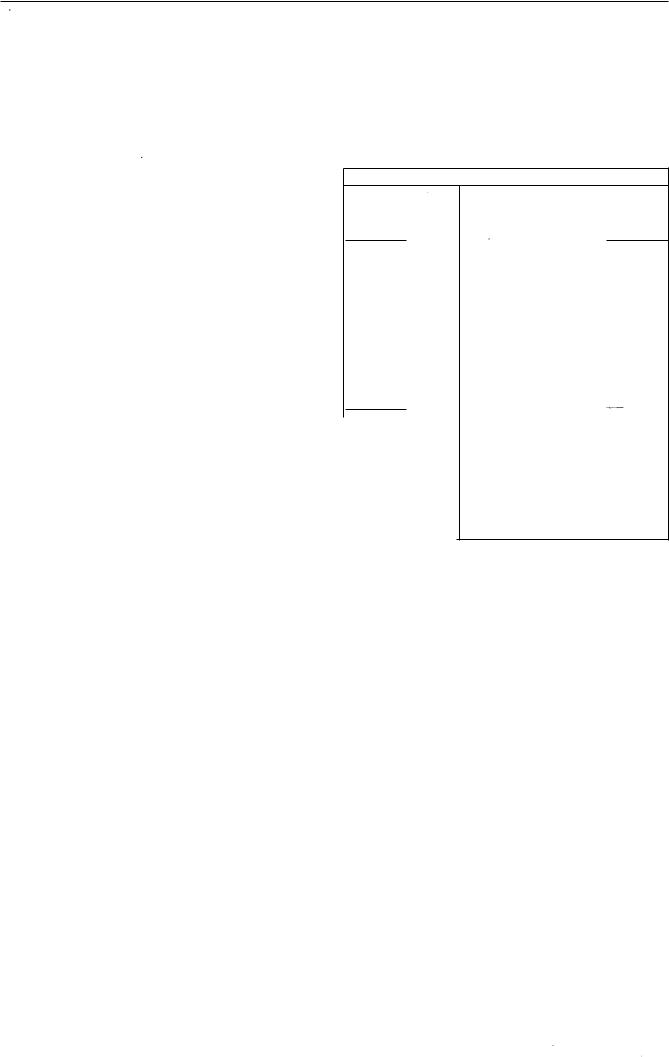
Температура:
|
|
Час (хв) |
Температура ес) |
|
|
|
|
|
|
|
:Колонка |
0 - 1 0 |
45 |
|
|
|
1 0 |
- 78 |
45 1 80 |
|
|
78 |
- 88 |
1 80 |
|
|
|
|
|
|
Блок вводу проб |
|
|
200 |
|
Детектор |
|
|
240 |
|
|
|
|
|
Детектор: полуменево-іонізаціЙниЙ.
Об'єм проби, що вводиться: 0.2 мкл.
Порядок виходу піків: має відповідати порядку зазна чення речовин ускладі розчину порівняння. Відміча ютьчаси утримування цих субстанцій.
Придатність хроматографічної системи: розчин по рівняння:
-коефіцієнт розділення: не менше 1 .5 для піків ліна-
лолу і -каріофілену.
Використовуючи часи утримування, визначені із хро маТ0грами розчину порівняння, визначаютьположен ня компонентів розчину порівняння на хроматограмі випробовуваного розчину.
Вміст компонентів, увідсотках, маєзнаходитисяута кихмежах:
- цинеол: менше 1 .0 %,
- ліналол: від 1 .5 % до 3.5 %,
-{З-каріофілен: від 1 .5 % до 7.0 %,
-сафрол: менше 3.0 %,
-транс-коричний альдегід: менше 3.0 %,
-цинамілацетат: менше 2.0 %,
-евгенол: від 70 % до 85 %,
-кумарин: менше 1 .0 %,
ЗБЕРІГАННЯ
У повітронепроникному максимально наповненому контейнері, у захищеному від світла та нагрівання місці.
КОФЕЇН
CofТeїnит
CAFFEINE |
|
|
' |
k ?" |
|||
"'С"і, |
, |
N:> |
|
|
І |
N |
|
O |
N |
|
|
CSHION402 |
снз |
М.м. 194.2 |
|
[56-08-2] |
|
|
|
474 |
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇН И 1 .2 |

Кофеїн
Кофеїн містить не менше 98.5 % і не більше іО і .5 % і,3,7-триметил-3,7-дигідро-lН-пурин-2,6-діону, у пе рерахунку на суху речовину.
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого "'або майже білого....кольоруабо шовковисті кристали білого "'або майже білого....кольору. Легко сублімується.
Розчинність. Помірно розчинний у воді Р, легко роз чинний у киплячій воді Р, малорозчинний в етанолі Р.
(Розчиняється в концентрованих розчинах бензоатів або саліцилатів лужних металів.)
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: А, В , Е. Друга ідентифікація: А, С, D, Е, F.
А. Температураплавлення (2.2. 14). Від 234 ос до239 ос.
В. Інфрачервоний спектр (2.2.24) субстанціїмає відпо відатиспектру ФСЗкофеїну.
С. До 2 мл насиченого розчину субстанції додають
0.05 мл розчину калію йодиду йодованого Р; розчин за лишається прозорим. Доодержаногорозчинудодають
0. 1 мл кислоти хлористоводневої розведеної Р; утво рюється коричневий осад, що розчиняється при ней тралізаціїрозчином натрію гідроксидурозведеним Р.
D. Близько 10 мг субстанції поміщають у пробірку із притертою скляною пробкою, розчиняють У 0.25 мл суміші 0.5 мл ацетилацетону Р і 5 мл розчину натрію гідроксидурозведеного Р. Одержаний розчин нагрівають у водяній бані при температурі 80 0С протягом 7 хв, охолоджують і додають 0.5 мл розчину дuметиламіно бензальдегіду Р2. Ще раз нагріваютьу водяній бані при температурі 80 ос протягом 7 хв, охолоджують і дода ють іО мл води Р; з'являється інтенсивне синє забарв лення.
Е. Субстанція має відповідати вимогам щодо втрати в масі при висушуванні, зазначеним у розділі «Випро бування на чистоту».
F. Субстанціядає реакцію на ксантини (2.3. 1).
ВИПРОБУВАННЯ НА ЧИСТОТУ
Розчин S. 0..5 г субстанції розчиняють при нагріванні у 50 мл води, вільноївід вуглецю діоксиду, Р, приготова ної з води дистильованої Р, охолоджують і доводять об'єм розчину тим самим розчинником до 50 мл.
Прозорість розчину (2.2. 1). Розчин S має бути прозо рим.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 1 .2
Кольоровість розчину (2.2.2, метод І/). Розчин S має бути безбарвним.
Кислотність. До 1О мл розчину S додають 0.05 мл роз чинубромтимолового синього РІ; з'являється зелене або жовте забарвлення, що переходить у синє при дода ванні не більше 0.2 мл 0.01 Мрозчину натрію гідрокси ду.
Супровідні домішки. Визначення проводять методом тонкошарової хроматографії (2.2.27), використовую чи як тонкий шар силікагель GF254 Р.
Випробовуваний розчин. 0.2 г субстанції розчиняють у сумішіметанол Р- ,..метиленхлорид Р....(4:6) ідоводять об'єм розчину тією самою сумішшю розчинників до
і О мл.
Розчин порівняння. 0.5 мл випробовуваного розчину доводять сумішшю метанол Р - ,..метиленхлорид Р....
(4:6) до об'єму іОО мл.
Налініюстарту хроматографічноїпластинки наносять 10 мкл (200 мкг) випробовуваного розчину і 10 мкл
(1 мкг) розчину порівняння. Пластинку поміщають у камеру із сумішшюрозчинниківрозчин аміаку концен трований Р - ацетон Р- ,..метиленхлорид р....- бутанол Р
(10:30:30:40). Коли фронт розчинників пройде і5 см відлініїстарту, пластинку виймають із камери, сушать на повітрі та переглядають в УФ-світлі за довжини хвилі 254 нм.
На хроматограмі випробовуваного розчину будь-яка пляма, крім основної, не має бути інтенсивнішою за пляму на хроматограмі розчину порівняння (0.5 % ) .
Сульфати (2.4. 13). Не більше 0.05 % (500 ррт). і5 мл розчину S мають витримувати випробування на суль фати. Еталон готують із використанням суміші 7.5 мл
еталонногорозчинусульфату (JOррт SO,J Рі 7.5 мл води дистильованої Р.
Важкі метали (2.4. 8, метод С). Не більше 0.002 %
(20 ррт). 1 .0 г субстанції має витримувати випробу вання на важкі метали. Еталон готуютьіз використан ням 2 мл еталонногорозчину свинцю (JOррт РЬ) Р.
Втрата в масі при висушуванні (2.2.32). Не більше 0.5 % .
і . ООО г субстанції сушать при температурі і05 о с про тягом 1 год.
Сульфатна зола (2.4. 14). Не більше 0.1 % . Визначення проводять із 1 .0 г субстанції.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0. і70 г субстанції розчиняють при нагріванні у 5 мл
кислоти оцтової безводної Р. Охолоджують, додають і О мл оцтового ангідриду Р, 20 мл толуолу Рі титрують 0. 1 М розчином кислоти хлорної потенціометрично
(2.2.20).
і мл 0. 1 Мрозчину кислоти хлорноївідповідає і 9.42 мг
CsHloN402·
475

Кофеїн моноrїдрат
ДОМІ Ш КИ
Сnецифіковані домішки: А.
Інші домішки, що виявляються: В, С.
А. теофілін,
|
|
о |
|
о |
H |
Нз ", |
|
|
|
||
С |
|
rNH |
|
||
|
N |
" |
JЗ |
- |
|
|
|
N |
|
NH2 |
|
oAСН.І. |
|
|
|||
В. N-[6-аміно- l ,3-диметил-2,4(lН,3Н)-діоксопіри
мідин-5-іл]формамід, |
:]:) |
||
|
|
||
нзе" |
|
о |
|
N |
|
N |
|
|
|
||
|
|
N |
|
|
|
І |
|
oAN |
\ |
||
|
|
СНЗ |
СНз |
С. 1 ,3,9-триметил-3,9-дигідро- l Н- пурин-2,6-діон (ізокофеїн)...tII
N
Речовини, що легко обвуглюються. 0.5 г субстанції роз чиняють у 5 мл кислоти сірчаної Р. Одержаний розчин має бути прозорим (2.2. 1) і безбарвним (2.2.2, метод 1).
•
КОФЕЇН МОНОГЩРАТ
Coffeinum monohydricum
CAFFEINE MONOHYDRATE
..tII
C81110 402,1I20 |
М.М. 212.2 |
[5743-12-4]
Кофеїн моногідратмістить не менше 98.5 % і не більше
101.5 % 1 ,3,7-триметил-3,7-дигідро-1 Н-пурин-2,6-діо ну, у перерахунку на суху речовину.
ВЛАСТИ ВОСТІ
Опис. Кристалічний порошок білого або майже біло го кольору або шовковисті кристали білого або майже білого кольору. Легко сублімується.
Розчинність. Помірно розчинний у воді Р, легко роз чинний у киплячій воді Р, мало розчинний уетанолі Р.
(Розчиняється в концентрованих розчинах бензоатів або саліцилатівлужних металів.)
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: А, В, Е. Друга ідентифікація: А, С, D, Е, F.
А. Температура плавлення (2.2. 14). Від 234 аСдо 239 ас Субстанцію сушать при температурі від 1 ОО аС до
105аС.
В. Інфрачервоний спектр (2.2.24) субстанціїмаєвідпо відати спектру Фе] кофеїну. Субстанцію сушать при
температурі від 100 ас до 105 ас
С. До 2 мл насиченого розчину субстанції додають
0.05 мл розчину калію йодиду йодованого Р; розчин за
лишається прозорим. До одержаного розчинудодають
0. 1 мл кислоти хлористоводневої розведеної Р; утво
рюється коричневий осад, що розчиняється при ней тралізаціїрозчином натрію гідроксидурозведеним Р.
D. Близько 1О мг субстанції поміщають у пробірку із
притертою скляною пробкою, розчиняють У 0.25 мл суміші 0.5 мл ацетилацетону Р і 5 мл розчину натрію гідроксидурозведеного Р. Одержаний розчин нагрівають
у водяній бані при температурі 80 аС протягом 7 хв, охолоджують і додають 0.5 мл розчину диметиламіно бензальдегіду Р2. Ще раз нагрівають у водяній бані при
температурі 80 аС протягом 7 хв, охолоджують і дода ють 1О мл води Р; з'являється інтенсивне синє забарв лення.
Е. Субстанція має відповідати вимогам щодо втрати в масі при висушуванні, зазначеним у розділі « Випро бування на чистоту» .
F. Субстанція дає реакцію на ксантини (2.3. 1).
ВИПРОБУВАННЯ НА Ч ИСТОТУ
Розчин S. 0.5 г субстанції розчиняють при нагріванні у 50 мл води, вільноївід вуглецю діоксиду, Р, приготова-
ної з води дистильованої Р, охолоджують і доводять об'єм розчину тим самим розчинником до 50 мл.
476 |
ДЕРЖАВНА ФАРМАКОП ЕЯ УКРАЇН И 1 .2 |

КунжутнаОJJія рафінована
Прозорість розчину (2.2. 1). Розчин S має бути прозо рим.
Кольоровість розчину (2.2.2, 'метод 11). Розчин S має бути безбарвним.
Кислотність. До 10 мл розчину S додають 0.05 млроз чинубро,Мти,Моловогосинього РІ; з'являється зелене або
жовте забарвлення, ЩО переходить У синє при дода ванні не більше 0.2 мл 0.01 Мрозчинунатрію гідрокси-
оо ду.
Супровідні домішки. Визначення проводять методом
тонкошарової хроматографії (2.2.27), використовую чи як тонкий шар силікагель GF254 Р.
Випробовуваний розчин. 0.2 г субстанції розчиняють у суміші ,Метанол Р - 'метиленхлорид Р (4:6) і доводять
об'єм розчину тією самою сумішшю розчинників до
10 мл.
Розчин порівняння. 0.5 мл випробовуваного розчину доводять сумішшю ,Метанол Р - 'метиленхлорид Р (4:6)
до об'єму 100 мл.
Налінію старту хроматографічної пластинки наносять 10 мкл (200 мкг) випробовуваного розчину і 10 мкл
( 1 мкг) розчину порівняння. Пластинку поміщають у камеру із сумішшю розчинниківрозчин а,Міаку концен трований Р - ацетон Р - 'метиленхлорид Р - бутанол Р
( І0:30:30:40). Коли фронт розчинників пройде 15 см відлінії старту, пластинку виймають із камери, сушать на повітрі та переглядають в УФ-світлі за довжини хвилі 254 нм.
На хроматограмі випробовуваного розчину будь-яка пляма, крім основної, не має бути інтенсивнішою за пляму на хроматограмі розчину порівняння (0.5 %).
Сульфати (2.4. 13). Не більше 0.05 % (500 ррт). 15 мл розчину S мають витримувати випробування на суль
фати. Еталон готують із використанням суміші 7.5 мл
еталонногорозчинусульфату (10ррт SO,J Рі 7.5 мл води дистильованої Р.
Важкі метали (2.4.8, 'метод С). Не більше 0.002 %
(20 ррт). 1 .0 г субстанції має витримувати випробу
вання на важкі метали. Еталон готуютьіз використан ням 2 мл еталонногорозчину свинцю (10ррт РЬ) P..iiJ
Втрата в масі при висушуванні (2.2.32). Від 5.0 % до
9.0 %. 1 .000 гсубстанціїсушать при температурі І05 аС протягом І год.
"'Сульфатна зола (2.4. 14). Не більше 0. 1 %. Визначен ня проводять із 1 .0 г субстанції.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0. 170 г субстанції, попередньо висушеної при темпе-
ратурі від 100 асдо 105 ас, розчиняють при нагріванні у 5 мл кислоти оцтової безводноїР, охолоджують, додають 10 мл оцтовогоангідриду Р, 20 мл толуолу Р і тит-
рують 0. 1 Мрозчино,М кислотихлорноїпотенціометрич но (2.2.20).
І мл 0. 1 Мрозчину кислоти хлорної відповідає 19.42 мг
CsHloN40z·
ДОМІШКИ
Сnецифіковані до,Мішки: А.
Інші дО'мішки, що виявляються: В, С.
А. теофілін,
В. N-[6-аміно- 1 ,3-диметил-2,4( І Н,3Н)-діоксопіримі дин-5-іл]формамід,
С. І ,З,9-триметил-3,9-дигідро- 1 Н-пурин-2,6-діон (ізокофеїн)..iiJ
•
КУНЖУТНА ОЛІЯ РАФІНОВАНА
Sesami оlеит raffinatum
SESAMEOIL, REFINED
Жирна олія, одержана зі стиглого насіння Sesamum indicum L. методом пресування або екстракції та потім
рафінована. Покращення кольорута запаху може бути досягнуто подальшим рафінуванням. Може бути до даний підхожий антиоксидант.
ВЛАСТИВОСТІ
Опис. Прозора, світло-жовтого кольору, майже безбарвна рідина.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАїН И 1 .2 |
477 |

Кунжутна олія рафінована
Розчинність. Практично не розчинна в 96 % сnирті Р,
змішується з петролейним ефіром Р.
(Відносна густина: близько 0.919.)
(Показник заломлення: близько 1 .473.)
(Застигає до м'якої маси при температурі близько
-4 0С)
ІДЕНТИФІКАЦІЯ
Перша ідентифікація:А. Друга ідентифікація: В.
А. Субстанція має відповідати вимогам щодо складу тригліцеридів, зазначеним у розділі « Випробування на чистоту».
В. Проводять ідентифікацію жирнихолій методом тон кошарової хроматографІЇ (2.3.2). Одержана хромато грама має бути порівнянною з типовою хроматогра мою кунжутноїолії.
ВИПРОБУВАН НЯ НА ЧИСТОТУ
Кислотне число (2.5. /). Не більше 0.5; визначення про водять із 10.0 г субстанції. Не більше 0.3, якщо суб-
станція призначена для виробництва лікарських за собів для парентерального застосування.
Перекисне число (2.5.5). Не більше 10.0. Не більше 5.0, якщо субстанція призначенадля виробництвалікарсь ких засобів для парентерального застосування.
Неомилювані речовини (2.5. 7). Не більше 2.0 %. Ви значення проводять із 5.0 г субстанції.
Лужні домішки (2.4. /9). Субстанція має витримувати випробування на лужні домішки в жирних оліях.
Бавовняна олія. 5 мл субстанції поміщають у пробірку та змішують із 5 мл суміші рівних об'ємів nентанолу Р
у розчину 10 г/л сірки Р у вуглецю дисульфіді Р. Одер жану суміш обережно нагрівають, доки не буде витіс
нений вуглецю дисульфід, і занурюють пробірку на однутретинутідовжини у киплячийрозчин натріюхло ридунасичений Р; протягом 15 хв не має з'являтися чер
воне забарвлення.
Склад тригліцеридів. Рідинна хроматографія (2.2.29).
Випробовуванийрозчин. 50.0 мг субстанціїдоводять су мішшю рівних об'ємів ацетону Р і метиленхлориду Р
до об'єму 10.0 мл.
58
10 12
з
7
ІЗ
14
|
|
11 |
17 |
|
|
__ |
I |
15 |
18 |
|
|
|
. 20 |
||||
_______________________ |
2 |
--J |
16 |
19 |
21 __ __ |
1 . ЛЛЛи |
4. |
о |
5 |
10 15 |
20 |
0ЛЛи |
|
7. |
ПЛЛ |
||
2. 0ЛиЛи |
5. |
0ЛЛ |
|
8. |
00Л |
З. ЛЛЛ |
6. |
00Лн |
|
9. |
СЛЛ |
25зо З5
10.ПОЛ І І . П ПЛ
12.000
40 |
45 |
50 |
|
І З. СОЛ |
|
|
14. |
ПОО |
|
15. |
ПСЛ |
55
60 |
хв |
19. сел |
16. |
П ПО |
|
17. СОО |
2. ппс |
|
18. ПСО |
21 . ССО |
|
Рисунок 0433.- 1. - Типова хроматограма, одержана у випробуванні «Склад тригліцеридів» оліїкунжутної рафінованої
478 |
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 1 .2 |
|

Кунжутна олія рафінована
Розчини порівняння: 80.0 мг триолеїну Р розчиняють у суміші рівних об'ємів ацетону Р і метиленхлориду Р і
доводять об'єм розчинутією самою сумішшю розчин никівдо 50.0 мл. Готують 5 розчинів порівняння роз веденням одержаного розчинутак, шобЇх концентрації знаходилися у межах: від невраховуваного мінімуму (0.5 %) до верхньої межі дЛЯ ОЛЛ (30.0 %).
Будують графік залежності логарифму плоші піка три олеїну від логарифму маси наважки триолеїну у роз чині порівняння, у міліграмах.
Колонка: 2 послідовно з'єднані колонки:
-розмір кожноїколонки: 0.25 м х 4 мм;
-нерухома фаза:силікагель октадецилсилільний дляхроматографії Р (5 мкм).
Рухома фаза:
-рухома фаза А: ацетон Р - метиленхлорид Р - ацето нітрил Р (5: 1 5:80);
-рухома фаза В: ацетон Р - метиленхлорид Р - ацето нітрил Р (20:20:60);
Час |
Рухома фаза |
А |
Рухома фаза |
В |
|||
(хв) |
(% |
06/06) |
(% |
06/06) |
|||
0 - 1 5 |
|
|
0 |
|
|||
|
|
1 00 75 |
|
25 |
|
||
|
|
|
|
|
|
|
|
1 5 |
- 25 |
|
75 |
|
|
25 |
|
|
|
|
|
|
|
|
|
25 |
- 70 |
75 O |
|
25 |
1 00 |
|
|
|
|
|
|
|
|||
70 - 75 |
O 100 |
|
1 00 0 |
|
|||
|
|
|
|
|
|
|
|
75 |
- 80 |
|
1 00 |
|
|
О |
|
Швидкістьрухомоїфази: 1 .0 мл/хв.
Детектування: із використанням детектора по світло розсіюванню, для якого наведені нижче параметри є підхожими. Якщо детектор має інші параметри, їх ко ригують таким чином, шоб виконувалися вимоги при датності хроматографічної системи.
- газ-носій: азот Р;
-швидкість потоку: 0.7 л/хв;
-температура випарника: 85 еС;
-температурарозпилювача: 45 ес.
Об'єм проби, що вводиться: 20 MКJI.
Ідентифікація піків: використовують хроматограми розчинів порівняння для ідентифікації піка триолеї ну; для ідентифікації інших піків використовують хро матограму, представлену на Рис. 0433.- 1 . Жирні кис лоти позначено: ліноленова (Лн ), лінолева (Л), олеїнова (О), пальмітинова (П), стеаринова (С).
Придатністьхроматографічноїсистеми: випробовува ний розчин:
-коефіцієнт розділення: не менше 1 .5 для піків ООО
(триолеїн) і СаЛ.
Використовуючи калібрувальну криву для розчинів порівняння, визначають вміст кожного піка, що має площу більшу площі піка невраховуваного мінімуму (0.5 %), у відсотках. Припускаючи, що сума площ усіх піків становить 100 %, методом внутрішньої нормалі зації визначають вміст кожного зі специфікованих нижче 8 тригліцеридів, у відсотках.
Склад тригліцеридів має бути таким:
-ЛЛЛ: від 7.0 % до 19.0 %,
-ОЛЛ: від 1 3.0 % до 30.0 %,
-ПЛЛ: від 5.0 % до 9.0 %,
-ааЛ: від 1 2.0 % до 23.0 %,
- ПОЛ: від6.0 % до 14.0 %,
-ООО: від 5.0 % до 14.0 %,
-СаЛ: від 2.0 % до 8.0 %,
-ПОО: від 2.0 % до 10.0 %.
Вода (2.5. 12). Не більше 0.05 %, якщо субстанція при значена для виробництва лікарських засобів для па рентерального застосування. Визначення проводять із 5.0 г субстанції напівмікрометодом.
ЗБЕРІГАННЯ
У повітронепроникному максимально наповненому контейнері, у захищеному від світла місці.
Я кщо субстанція призначена для виробництва лікарських засобівдля парентерального застосування, її зберігають в атмосфері інертного газу у повітронеп роникному контейнері.
Після відкривання контейнера його вміст використо вують якнайшвидше. Будь-яка частка вмісту контей нера, що не використовується відразу, має зберігатися в атмосфері інертного газу.
МАРКУВАННЯ Зазначають:
-метод, яким одержана олія: пресуванням або мето дом екстракції,
якщо необхідно:
-субстанція придатна для виробництва лікарських засобів для парентерального застосування,
-назву використовуваного інертного газу.
__1V
Допускається використання зазначеноїнижче методи ки.
Складтригліцеридів. Випробування проводять методом рідинної хроматографії (2.2.29).
Випробовуванийрозчин. 0.200 г субстанції поміщають у мірну колбу місткістю 10 мл і доводять об'єм розчину рухомою фазою до 10.0 мл.
Хроматографування проводять на рідинному хрома тографі з рефрактометричним детектором за таких умов:
-дві послідовно з'єднані колонки із нержавіючоїсталі розміром 0.25 м х 4.6 мм, заповнені силікагелем ок тадецилсилільним для хроматографії Р із розміром частинок 5 мкм,
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇН И 1 .2
479

Кунжутна олія рафінована
ОЛнЛн
ЛЛЛн
ОЛЛ
ООЛ
ЛЛЛ
ООО
ООЛн ОЛЛн ПЛЛ СЛЛ ПОЛ
СОЛ ПОО |
СОО |
||
|
|
|
|
о |
1 0 |
30 |
50 |
60 |
|
|
|
|
хв |
Рисунок. - Типова хроматограма, одержана у випробуванні «Склад тригліцеридів» оліїкунжутноїрафінованої
-рухома фаза: метиленхлорид Р - ацетонітрил Р ( 1 :2),
-швидкість рухомої фази 1 .0 мл/хв.
Хроматографують 20 мкл випробовуваного розчину. Ідентифікують піки, використовуючи типову хрома тограму (див. Рисунок). Залишки жирних кислот по-
значено: ліноленової (Лн), лінолевої(Л), олеїнової (О), пальмітинової (П), стеаринової (С).
Визначають вміст тригліцеридів, у відсотках, із площ піків на хроматограмі випробовуваного розчину мето дом внутрішньої нормалізації.
480 |
ДЕРЖАВНА ФАРМАКОП ЕЯ УКРАЇНИ 1 .2 |
Лавандова олія
л
ЛАВАНДОВА ОЛІЯ
Lavandulae aetheroleum
LAVANDER OIL
Ефірна олія, одержана із квітучих верхівок Lavandula angusti/olia Мillег (Lavandula officinalis Chaix) методом
перегонки з водяною парою.
ВЛАСТИВОСТІ
Опис. Прозора, безбарвна або блідо-жовтого кольору рідина.
(Субстанція має характерний запах.)
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: В. Друга ідентифікація: А.
А. Тонкошарова хроматографія (2.2.27).
Випробовуванийрозчин. 20 мкл субстанції розчиняють в 1 мл толуолу Р.
Розчин порівняння. 10 мклліналолу Рі 10 мклліналіл аце тату Ррозчиняють в 1 мл толуолу Р.
Пластинка: тшхпластинка із шаром силікагелю Р.
Рухома фаза: етилацетат Р - толуол Р (5:95).
Об'єм проби, що наноситься: 10 мкл, смугами.
Відстань, яку має пройти рухома фаза: І О см від лінії старту, двічі з інтервалом у 5 хв.
Висушування: на повітрі.
Виявлення: обприскуютьрозчином анісового альдегіду Р,
нагрівають при температурі від 1ОО ОС до 105 ОС про тягом 5- 1 О хв і відразу переглядають при денному світлі.
Результати: нижче наведено послідовність зон на хро матограмах розчину порівняння та випробовуваного розчину. На хроматограмі випробовуваного розчину можуть виявлятися інші зони фіолетово-червоного або зеленувато коричневого кольору вище зони ліналіл ацетату, безпосередньо близько фронту розчинників.
Верхня частина пластинки
декілька фіолетово-червоних або зеленувато-коричневих зон
ліналілу ацетат; зона
'від фіолетового до
коричневого кольору
|
|
|
зона від фіолетового до коричневого |
|
|
|
|
|
кольору (ліналіл ацетат) |
|
|
|
|
|
фіолетово-червона зона |
|
|
|
|
|
можлива наявність слабко забарвленої |
|
|
|
|
|
фіолетово-коричневої зони (цинеол) |
|
|
, |
. |
. |
|
|
|
зона від фіолетового до коричневого |
|
||||
[ |
ЛІналол; зона вІД |
|
|||
фіолетового до |
кольору (ліналол) |
|
|||
|
коричневого кольору |
|
|
|
|
|
|
|
слабко забарвлена |
|
|
|
|
|
фіолетово-коричнева зона |
|
|
|
|
|
декілька зон невизначуваних речовин |
|
|
|
|
|
|
||
|
Розчин порівняння |
Випробовуваний розчин |
|
||
В. Переглядають хроматограму, одержану у випробу ванні на хроматографічний профіль.
Нормування: часи утримування характерних піків на хроматограмі випробовуваного розчину мають співпа дати із характерними піками на хроматограмі розчину порівняння (а).
ВИПРОБУВАННЯ НАЧ ИСТОТУ
Відносна густина (2.2.5). Від 0.878 до 0.892.
Показник заломлення (2.2.6). Від 1 .455 до 1 .466.
Оптичне обертання (2.2. 7). Від -12.50 до -7.00.
Кислотне число (2.5. І). Не більше 1 .0. 5.0 г субстанції розчиняють у 50 мл зазначеної суміші розчинників.
Хроматографічний профіль. Газова хроматографія
(2.2.28): метод внутрішньої нормалізації.
Випробовуванийрозчин. Випробовувана субстанція.
Розчин порівняння (а). 0. 1 г лімонену Р, 0.2 г цинеолу Р,
0.2 г 3-0ктанону Р, 0.05 г камфори Р, 0.4 г ліналолу Р,
0.6 гліналілуацетату Р, 0.2 г терnінен-4-0ЛУР, 0. 1 гла вандололу ацетату Р, 0.2 г лавандололу Р і 0.2 г а-тер nінеолу Р розчиняють у 5 мл гексану Р.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇН И 1 .2 |
48 1 |