Fig. 16.6. LHC II vibrational density of states (a), and the latter divided by ω2 (b), from NEAT experiments; data from [41]
seem to generally reflect the disordered nature of protein molecules. The wide distribution of protein vibrations found for LHC II can be understood in terms of structurally inequivalent protein domains within the LHC II trimer leading to a partial localization of protein phonons. One possible interpretation is, that the low-energy vibrational peak of LHC II at about 2.3 meV corresponds to vibrations fully delocalized over the protein matrix of the LHC II monomer or trimer. In contrast to this, the slight tailing towards higher vibrational frequencies may be constituted by separate contributions from vibrations with smaller amplitudes. These vibrations might e.g., be localized on one of the di erent α-helices forming the protein matrix of LHC II.
16.4 Non-Gaussian Motion
It must be pointed out that there are many situations where Gaussian behavior of scattering functions is absent. Because of strongly non-Gaussian (spaceand time-dependent) atomic probability–density distributions, the Gaussian approximation is then either far from being applicable, or limited to purely phenomenological descriptions.4 This is probably true in the case of many
4These may nevertheless be useful for some practical purposes
368 R.E. Lechner et al.
of the functionally most relevant motions of molecular subunits in biological macromolecules exhibiting large anisotropic atomic displacements. Here are a few of the most simple examples of models of non-Gaussian motion.
16.4.1 Atomic Jump Motions Described by Rate Equations
The simple di usion theory is satisfactory for our purpose only, when the locations (r, t) in space and time, where experimental observations are made, are at distances from the space-time origin, that are large as compared to the small elementary di usive step distance l and the residence time τ0 between consecutive steps. If, however, such small values of (r, t) are to be covered by the experiment, the simple-di usion description breaks down, and Ss(Q, ω) then significantly di ers from the above case, because it contains structural information concerning the details of the local geometry directly related to the interatomic distances and the spatial arrangement of atoms.
A model was proposed by Chudley and Elliott [12], to obtain the classical self-correlation function Gs(r, t) for an atom di using on an assumed quasicrystalline lattice of a liquid. Because of the assumed regular spatial arrangement of atom sites and its relative simplicity, this so-called CE-model has also become very popular for treating atomic di usion on interstitial lattices in crystals. We briefly discuss it in the following, in order to provide some insight into how jump models based on rate equations are constructed.
Let us first consider di usion by atomic jumps on a Bravais lattice. One calculates the probability P (rm, t) to find the di using atom on a site rm of the lattice at time t, where it spends a time τ on the average. The time τj required for the di usive jump from site to site is neglected. The jumps occur between a given site rm and its neighbors rm + dν , where dν (ν = 1, 2, . . . , s) is a set of jump vectors connecting the given site with the neighbors. The master equation for P (rm, t) is then
s
∂P (rm, t)
1
1
= −
P (rm, t) +
ν=1 P (rm + dν , t) .
(16.20)
∂t
τ
sτ
The first and second term on the right side are the loss and growth rate due to the jumps to and from adjacent sites, respectively. In order to obtain Gs(r, t), the initial condition
P (rm, 0) = δ(rm)
(16.21)
is imposed. Because in a Bravais lattice all sites are equivalent, one directly obtains the self-correlation function as the solution of the master equation: Gs(r, t) ≡ P (rm, t). Several further assumptions made, when deriving Eq. 16.20 are that consecutive jumps are uncorrelated, and that blocking and mutual interaction e ects are neglected. This implies a low site occupancy c for the di using particles. The basic CE-model (represented by Eq. 16.20) has been extended to cover many special and more complex situations, for instance to include e ects due to atom–atom correlations [49, 50]. A general
16 Quasielastic Neutron Scattering: Applications
369
theory of the master equation is given in Chapter 18 of [51]. Correlated jumps are discussed in Chapters 1, 3, 10, and 18 of [51]. Now we introduce the Fourier
transform
+∞
P (rm, t) = P˜(Q, t)e−iQrm dQ,
(16.22)
−∞
˜ ≡
where P (Q, t) Is(Q, t) from Eq. 15.9, Sect. 15.2.1 in Part I, this volume. This leads to a di erential equation of first order for Is(Q, t) with an exponential decay function as solution,
Is(Q, t) = e−Γ (Q)|t| ,
(16.23)
which fulfils the initial condition Eq. 16.21. From this one gets
s
1
Γ (Q) =
(1 − e−iQdν ) .
(16.24)
sτ
ν=1
The resulting dynamic structure factor is the Fourier transform of IS(Q, t), i.e. the normalized Lorentzian
Ss(Q, ω) =
Γ (Q)/π
(16.25)
ω2 + Γ (Q)2
with energy halfwidth (HWHM) Γ (Q). For small Q, i.e., for Q 1/d, where d = length of jump vector, one gets the limiting case
Γ (Q) =
d2
6τ Q2 = DsQ2 .
(16.26)
i.e., the Gaussian behavior of Ss(Q, ω) and Is(Q, t) is recovered in this limit (see Eq. 16.4). This relation holds generally for jump di usion in three dimensions, and it is independent of the detailed jump geometry. When di usion occurs in a crystalline lattice, Γ (Q) is periodic in reciprocal space. It has a maximum at the Brillouin zone boundary and it is zero, if a reciprocal lattice point G is reached, such that Γ (Q = G) = 0. This “line narrowing”5 is related to Bragg di raction of the neutron wave from the probability density of the proton distributed over the sites of the Bravais lattice (see, for instance, [53–55]).
Finally, we mention here a useful simplification of expression 16.25, which has often been applied in the case of polycrystalline samples requiring orientational averaging of the theoretical scattering function. If, instead of Ss(Q, ω), the width function is averaged, one obtains the “isotropic approximation”:
Γ (Q) =
1
sin(Qr)
τ
1 −
Qr
(16.27)
5This e ect should not be confused with the coherent line narrowing predicted by De Gennes [52]
370 R.E. Lechner et al.
This relation can be used to determine an estimate of an (average) jumpdistance r from the Q-dependence of measured quasielastic line widths [7–9]. An example, where this procedure has been applied to two-dimensional di u- sion on the surface of a membrane, is discussed in Sect. 16.5.1 (see Fig. 16.14).
16.4.2 Confined or Localized Di usive Atomic and Molecular Motions
Di usive motions of individual atoms or molecules do not necessarily extend over the whole volume of the system under investigation. They may instead be restricted to a certain space of molecular order of magnitude. This occurs for instance, when atoms bound in a molecule – e.g., in a mobile sidegroup of a polypeptide chain – are undergoing rotational di usion with the latter, or when molecules are confined to di usion within the space available inside the cages of an inclusion compound. In the context of biological macromolecules, this situation may be encountered for instance by water molecules trapped inside of these molecules, or in cavities or grooves at their surface. Here, the dynamical problem consists in solving a suitable equation of motion applying the appropriate boundary conditions which correspond to the specific spatial limitation of the motion. Then, for instance, when a molecule or molecular subunit performs discrete reorientational steps (rotational jumps) – inspite of the spatial limitation – a rate equation of the type represented by Eq. 16.20 can still be employed, whereas for continuous di usion Eq. 16.6 may still apply. All molecular motions limited in space have in common, that the atomic probability density within the confinement does not decay to zero at infinite time. As a consequence, the temporal Fourier transform to be calculated for obtaining the Van Hove incoherent scattering function, contains an (elastic) δ(ω)-term, in addition to nonelastic terms (see Sect. 15.2.2 in Part I, this volume). Generally, the above type of equations for localized or confined diffusive motions yield scattering functions (here for convenience labelled with the subscript for rotation) of the following form [5] and [6]:
n
j
(16.28)
Srot(Q, ω) = A0(Q)δ(ω) + Aj (Q)Lj (Hj , ω).
=1
Here A0(Q) is the EISF of the localized di usive motion and the functions Aj (Q) are the n corresponding QISFs of the n Lorentzian components, Lj (Hj , ω), with halfwidths (HWHM) Hj , and with (see Sect. 15.2.2 and Fig. 15.1 in Part I, this volume):
n
n
j
(16.29)
QISF = QISFj (Q) =
Aj (Q) = 1 − EISF.
=1
j=1
Many models yielding expressions of the type 16.28 have been developed in the past for specific boundary conditions imposed by the geometry of di erent localized di usive motions. Several representative examples have been given in Table 15.1 and Fig. 15.1 of Part I, this volume, including the corresponding
16 Quasielastic Neutron Scattering: Applications
371
references to the original literature. When, in addition to the confined di usive motion, other dynamical phenomena, such as vibrational motions or translational di usion, are present in the observation time window of the experiment, Srot(Q, ω) may be combined with the pertinent additional scattering functions by convolution according to Eq. 16.2. Without going into a detailed discussion, we merely note here, that Eqs. 16.28 and 16.29 have been applied to isolate the contribution due to two-site and three-site jump-reorientation of small molecular subunits, such as methyl-groups and short side-chains to the QENS intensity observed in experiments on purple membrane [56]. The classical model of di usion on the surface of a sphere [57] (corresponding, e.g., to continuous rotational di usion of molecules in solution) has been formulated for incoherent, as well as for coherent scattering, and applied not only for the former [58–62] (see also [1] and [3]), but also in the latter case [61, 63, 64]. The model for continuous di usion in the interior of a spherical volume [65] has become popular for the description of localized di usive (confined molecular) motion in the context of protein dynamics [66–72]. H-bond flip-flop motions based on the assumption of a double-well potential have been treated using a symmetric two-site jump model [73]. A potentially interesting model for dealing with H-bond formation–breaking reaction equilibria in biological macromolecule/hydration–water complexes, which is based on an asymmetric two-site jump model, can be found in [74].
16.4.3 Environment-Dependence
of Confined Di usive Protein Motions:
Example Lysozyme
Any functional activity of biological systems involves the quasicontinuous performance of multiple conformational rearrangements of macromolecules such as proteins, enzymes, etc. [75]. The molecular dynamics involved is fully developed only at physiological conditions of the environment. Because of the strong correlation between functionality, dynamics and environment, it is of great interest, to study the dynamics of biological systems as a function of environmental parameters. These may concern physical or biochemical aspects, as temperature, pressure,..., humidity, salinity,..., and – more generally – the kind and concentration of molecular and ionic species surrounding the macromolecular surface. It is well-known that the omnipresence of liquid water in biological systems has a plasticizing e ect ensuring the conformational flexibility (e.g., of proteins) required for biological function (see also Sect. 16.5.2). On the other hand, proteins embedded in sugars or glycerol have higher melting temperatures, i.e., they possess an increased thermostability, as compared to the same systems in a state of hydration. The reason is, that under certain conditions well above room temperature, proteins are still stable in anhydrous solvents, when the plasticizing action of water which would give them a conformational flexibility su cient for denaturing in the hydrated state, is absent [76]. In general, thermal fluctuations of proteins, and in particular stochastic motions occurring on the picoto nano-second time-scales
372 R.E. Lechner et al.
accessible to neutron scattering, are a ected by the character of the environment. Neutron scattering experiments have been carried out, employing the elastic-window scan technique [77] (see Sect. 15.3.3 in Part I, this volume), for studying the molecular mobility of lysozyme solvated in glycerol, with various water contents, as a function of temperature [78, 79]. These studies have revealed the alteration of the protein’s internal mobility, when the character of its environment changes from a stabilizer-like to a plasticizer-like nature. Addition of water strongly a ects the dynamical behavior of glycerol-solvated lysozyme, which with this technique is monitored via a measured e ective mean square displacement of hydrogen atoms in the protein. A quasielastic neutron scattering experiment shows this e ect in more detail [39]. As an example, Fig. 16.7 shows IINS spectra of dry lysozyme solvated in glycerol, as obtained at Q = 1.1 ˚A−1, at 100 K (open circles) and at 300 K (full circles). The low-Tspectrum essentially exhibits a broad Boson-peak type component centered near 4 meV, which is due to quasiharmonic vibrations. There is also a very small quasielastic contribution at energy transfers below about 2 meV. At 300 K, where di usive motions have become rather important, a large quasielastic component dominates the spectral shape. The continuous line is the elastic energy resolution function measured with a vanadium plate. The inset shows the same spectra, but rescaled by dividing through the Bose
Intensity (a.u.)
0.04
0.03
0.02
0.01
0.00
(a.u.)
0.03
0.01
Intensity
0.02
0.00
0
4
8
12
E (meV)
0
4
8
12
E (meV)
Fig. 16.7. Inelastic neutron scattering spectra of dry lysozyme solvated in glycerol, at 100 K (open circles) and 300 K (full circles), at Q = 1.1 ˚A−1, measured with the MTOF spectrometer NEAT; see Sect. 15.3.2 in Part I, this volume. The continuous line is the elastic energy resolution function measured with a vanadium plate. Inset: for energy transfers above 5 meV, where the quasielastic component is negligible, the inelastic region of the 100 K spectrum overlaps precisely with that of the 300 K spectrum, after both have been rescaled by dividing through the Bose phononpopulation factor (after [39])
16 Quasielastic Neutron Scattering: Applications
373
phonon-population factor, in order to make the vibrational part of the scattered intensity temperature-independent. The result of this procedure is, that the vibrational components observed at the two di erent temperatures are perfectly overlapping in the energy transfer region above 5 meV, where the (quasielastic) spectral contribution due to di usive motions is negligible. The spectral weight of the quasielastic component varies strongly not only with the temperature, but also with the degree of hydration. This is shown in Fig. 16.8, where spectra of lysozyme in glycerol at 300 K are presented for three di erent hydration levels: 0.0h (open circles), 0.2h (full circles), 0.4h (open squares). One observes that, similarly to the temperature e ect, increased hydration induces an increased global mobility of lysozyme: both, the inelastic and the quasielastic signals are increased at higher hydration levels. This e ect is particularly large in the latter case. In order to investigate the nature of the relaxational dynamics, the quasielastic spectral shape must be studied alone. The vibrational component, as determined at 100 K (see Figs. 16.7 and 16.8), can be fitted by a suitable function, like the phonon-expansion described in Sects. 16.3.3 and 16.3.4 (see Eqs. 16.10–16.16). When this is subtracted from the measured spectra, the sole quasielastic component remains. This is shown in Fig. 16.9 for the room temperature (300 K) spectra at the three hydration levels, 0.0h (open circles), 0.2h (full circles) and 0.4h (open squares). These spectra were quantitatively analyzed by fitting a quasielastic scattering function of the type 16.28, folded with the experimental resolution function. Two Lorentzian components were found to well reproduce the spectra, with the QISFs (Aj (Q)), the linewidths (Hj ) and a trivial global normalization
Intensity (a.u.)
0.06
0.04
0.02
0.00
0
4
8
E (meV)
Fig. 16.8. Inelastic neutron scattering spectra of lysozyme solvated in glycerol, at room temperature (300 K), for three di erent hydration levels, 0.0h (open circles), 0.2h (full circles) and 0.4h (open squares), measured with the MTOF spectrometer NEAT. The momentum transfer value is Q = 1.1 ˚A−1 (after [39])
374 R.E. Lechner et al.
Intensity (a.u.)
0.15
0.10
0.05
0.00
−1
0
1
E (meV)
Fig. 16.9. Quasielastic neutron scattering spectra of lysozyme solvated in glycerol for a momentum transfer value of Q = 1.1 ˚A−1, at room temperature (300 K), for three di erent hydration levels, 0.0h (open circles), 0.2h (full circles) and 0.4h (open squares), measured with the MTOF spectrometer NEAT. The vibrational component of the scattered intensity has been subtracted (see text). The narrow continuous line is the elastic energy resolution (after [39])
factor as free parameters. Figure 16.10 shows the fit result for the 0.4h sample. It is seen that the two Lorentzians exhibit rather di erent widths, namely (H1 = 0.135 ±0.010 meV) and (H2 = 2.45 ±0.70 meV), which means that two relaxational phenomena occurring on quite di erent time scales are observed. For di erent values of the hydration (h), the same pair of widths is found for the two Lorentzians, but the quasielastic signals (i.e., the QISFs) increase with h. This is the signature of a progressive onset of new relaxational degrees of freedom of internal protein motions, probably related to groups located at the protein surface. Apparently, also in the presence of glycerol, water molecules are able to gradually activate the whole lysozyme dynamics by progressively hydrating the protein surface groups. This may be interpreted as a hydrationdriven onset of confined di usive internal protein motions.
16.4.4 Change of Protein Dynamics on Ligand Binding: Example Dihydrofolate Reductase
It is of fundamental importance in biology and medicine to understand how ligands bind to proteins [80–85]. Various types of interactions may be involved in ligand association: van der Waals, electrostatic and hydrophobic interaction, hydrogen bonding, or di erent intermolecular interactions specific for the protein–ligand complex. The signature of complex-formation is expected to be visible in the spectrum of protein vibrations or – more
16 Quasielastic Neutron Scattering: Applications
375
Intensity (a.u.)
10
1
0.1
0.01
10
1
0.1
0.01
0 5 10 E (meV)
−1.0
−0.5
0.0
0.5
1.0
1.5
E (meV)
Fig. 16.10. Fit (full line) of a scattering function comprising an elastic and two quasielastic (Lorentzian) terms of di erent widths to the room temperature quasielastic spectrum of lysozyme solvated in glycerol, and hydrated at 0.4h (open circles), at Q = 1.1 ˚A−1. The dashed and dotted lines are the narrow and the broad Lorentzian components, respectively. The inset shows the quasielastic spectrum over a larger energy range. These spectra (after [39]) were derived from measurements with the MTOF spectrometer NEAT; see Sect. 15.3.2 in Part I, this volume
generally – of protein dynamics, as compared to that of the unbound protein [86–92]. An experimental investigation of the dynamic structure factor modification due to ligand binding was carried out using neutron scattering [93] on the enzyme dihydrofolate reductase (DHFR), an important target for anticancer and antibacterial drugs [94–98]. In the presence of DHFR and of the nicotinamide adenine dinucleotide phosphate (NADPH) cofactor, a catalytic reduction of dihydrofolate to tetrahydrofolate takes place. The ligand used is methotrexate (MTX), a folate antagonist of DHFR employed e ectively as a cytotoxic agent in the treatment of cancers [99]. In order to minimize scattering from solvent molecules, the system was exchanged with D2O. Both the uncomplexed enzyme (DHFR+NADPH) and the complexed enzyme (DHFR+NADPH+MTX) were hydrated to a degree of 30%, i.e., 30 mg of D2O per 100 mg of dry weight protein. It is important to keep in mind, that this degree of hydration is only valid for the environmental conditions under which it was established, whereas at much lower temperatures, part of the water will be e ectively separated from the protein due to a “dehydration by cooling” e ect [100], but might still be present within the sample container in the form of crystalline D2O-ice. But, because of the comparatively low scattering cross-section of D2O, the latter (with its 23% fraction of the total sample weight) will not contribute more than about 2.5% to the integral of the inelastic scattering spectrum.
376 R.E. Lechner et al.
5
5
.units)
4
4
(arb
3
3
−4
w)*10
2
2
10
20
30
40
50
S(q,
1
0
0
50
100
150
200
w (cm−1)
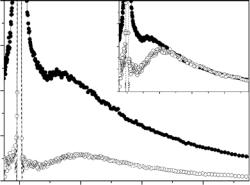
Fig. 16.11. Dynamic structure factor versus frequency ω [cm−1] for uncomplexed DHFR (lower curve) and complexed with methotrexate form of DHFR (upper curve) at 120 K. Data, taken with the direct-geometry TOF spectrometer IN6 (see Sect. 15.3.1 in Part I, this volume) using an incident neutron wavelength of 5.12 ˚A, have been summed over all scattering angles of the experiment (elastic Q-range approximately from 0.3 to 2.2 ˚A−1). Both spectra are normalized to the elastic peak height. Inset: low frequency region of the spectra; after [93]
Measurements were performed with the XTL-TOF spectrometer IN6 at ILL Grenoble (see Sect. 15.3.1 in Part I, this volume), at the fairly low temperature of 120 K, in order to minimize the contribution of di usive motions. Figure 16.11 shows the measured incoherent scattering functions of the uncomplexed and the complexed form of the enzyme. Both spectra exhibit, in a qualitatively similar manner, two fundamentally di erent parts: The first one is an inelastic region including the Boson peak (ω above 20 cm−1)6, which is probably exclusively due to harmonic vibrations. The second part is the lowfrequency region (ω below 20 cm−1), where a careful comparison with the energy resolution function determined by scattering from a vanadium sample (not shown in the figure) suggests that the harmonic vibrational component is overlapping with a quasielastic contribution on top of it. The latter indicates, that at 120 K the spectra still contain a non-negligible e ect from strongly anharmonic, or overdamped vibrational, or even from localized di usive modes of motion, inspite of the low temperature. This is, however, not unusual and has also been found earlier, for instance in the cases of hydrated purple membrane [101, 102], and in alanine dipeptide [103], where anharmonic motions and rotational motions of small side-groups have been observed down to 100 K and below (see also the 100 K spectra of Fig. 16.7 in Sect. 16.4.3). In this lowenergy region, a rather significant di erence was observed between the spectra of the complexed and the uncomplexed enzyme. The quasielastic component
6Note that the energy transfer unit 1 meV corresponds to 8.07 cm−1