

Neutron Scattering in Biology - Fitter Gutberlet and Katsaras
.pdf428 U. Lehnert et al.
C-terminus
CP
|
|
B |
|
E |
|
G |
|
D |
C |
||
|
|||
|
A |
||
|
|
||
|
F |
|
EC |
N-terminus |
|
D E |
B |
C |
F |
|
A |
G |
|
Fig. 18.3. Schematic view of selectively labeled BR in the PM and the projection structure of a BR trimer in the PM lattice. The entire PM has been deuterated, except for the Met and Trp residues and the chromophore retinal (shown in ball-and-stick mode) that have been kept hydrogenated. Structural data are from PDB entry 1qhj [38] and the figure has been prepared with MOLSCRIPT [39] and RASTER3D [40]
18.3.4 Lipid Environment
A point of discussion is always to which extent lipid motions contribute to the observed PM motions. Using partly delipidated PM samples, which are still crystalline and containing BR trimers, Fitter and coworkers [37] started addressing the role of the lipid environment on PM dynamics. A reduced flexibility of the delipidated membrane has been observed and the di erence in flexibility was more pronounced for a wet than for a dry membrane. This has led the authors to the conclusion that it is the hydration of the lipids which plays a major role for the dynamical behavior of the global membrane as suggested by Zaccai [23]. In addition, photocycle kinetics is partly slowed down upon delipidation and a correlation between the BR photocycle and the dynamical behavior of the protein–lipid complex in di erent conditions has been made [37].
A comparison between motions explored by quasielastic neutron scattering on PM and disk membranes, which contain rhodopsin and lipids in a weight ratio of 45% rhodopsin and 50% of lipids, indicated higher internal flexibility of the disk membranes [41]. From this piece of work a suggestion of a linear dependence between the amplitudes of motions (in this case extracted from
18 Relating Protein Dynamics to Function and Structure |
429 |
a two-site jump di usion model or di usion inside a sphere model) and the relative amount of lipids has been emerged.
What might be the impact of packing in the membrane on protein and lipid flexibility? Packing can be perceived as a lateral pressure on the protein, which maintains the molecule in a certain conformational state. It is important to note that BR is not inserted in a symmetric way in PM, but that the extracellular part of the protein is more deeply buried in the lipids. Therefore, the lateral pressure might not be identical on both sides of the protein. This agrees with the more rigid dynamics of the specifically labeled PM sample presented above. From neutron di raction experiments it has been concluded that BR needs a soft environment in order to be functional, which allows large conformational changes of the protein. In dry membranes or at low temperature, conditions in which BR is not functional, the close-packed environment would inhibit the required amplitudes for accomplishing the photocycle and the pump activity [23]. It has been pointed out that thermal dynamics serves as a kind of lubricant to enable large-scale displacements necessary to ensure correct protein activity [19, 25, 42]. Thus, it can be suggested that protein packing in the membrane participates in the creation of the necessary soft environment including the required well-defined rigidity to maintain the dynamical heterogeneity, which influences functionally important conformational changes during the photocycle. The di erent kinetics of the rise and decay of the photointermediates in monomeric BR compared to BR in PM have been attributed to the packing of BR in PM [43]. However, one has to be careful, because it is di cult to estimate which of the changed parameters such as monomerisation or disruption of lipid protein interaction produce the most prominent perturbation in the photocycle and of proton pumping. For lactose permease from E. coli, it has been suggested that the critical lateral lipid packing pressure is an essential prerequisite for correct membrane insertion and function of the protein [44]. The permease seems to be an extremely flexible, metastable molecule that requires external stabilization by the surrounding membrane and, therefore, it is di cult to crystallize such a membrane protein without its lipids. Recently, the first high-resolution structure became available [45].
18.3.5 Relation between PM Dynamics and Crystallographic B-factors
Electron and X-ray di raction data of BR provide temperature-factors (B- factors) as one result of the structure refinement process, which reflect both static and dynamic disorder. They have indicated that the protein is most rigid in its center near the retinal binding pocket and that it becomes more flexible at the surfaces [46]. Furthermore, it has been found that the temperaturefactors are asymmetrically distributed over the protein, i.e., larger in the cytoplasmic (CP) than in the extracellular (EC) region [38], supporting the neutron scattering results from R´eat et al. [19] discussed in Sect. 18.3.3. In
430 U. Lehnert et al.
molecular dynamics simulations, the fluctuations of the helix extremities in the CP half of BR appeared twice as large than those in the EC half [47].
The question of the physical significance of these temperature factors, and how they are related to the thermal motions observed in neutron scattering, can be addressed. The isotropic temperature-factor or B-factor is described by B = 8π2 <x2> with a mean square deviation <x2> from a mean atomic position. The overall <x2> of an atom originates from several sources including the internal static disorder (di erent configurations in di erent unit cells), internal dynamic disorder (vibrations within molecules), lattice defects and lattice vibrations. In the case of the BR ground-state structure determined at 100 K, the average <x2> value is 0.3 ˚A2 (see for example the B-factors from the PDB entry codes 1c3w [48] and 1cwq [49]). They are about four times larger compared to <u2> values from neutron scattering at 100 K (<u2> values are around 0.07 ˚A2). The motions, which enter in the B-factor calculation, are assigned to single atoms and reflect the vibrations of backbone and sidechain atoms. Vibrations of bonded atoms are of the order of 0.1 ˚A in a high frequency range (1013–1014 s−1) [1], whereas the <u2> reflect the motions of the amino acid side-chains (obtained from the scattering of the H-atoms in these groups) in a frequency range of 109–1012 s−1 [2, 7]. In contrast to the mean square amplitudes <u2>, the B-factors are not directly measured and only arise from the structure refinement. They mainly reflect a general tendency of the protein flexibility. They do not supply quantitative information in contrast to neutron scattering, which provided evidence for the dynamical heterogeneity. A comparison between temperature factors and root mean square deviation (RMSD) from an averaged BR structure of six models has revealed that the RMSD are mostly lower in the helices than the corresponding B-factors whereas the RMSD loop values are mostly higher. This has led to the suggestion that the B-factors are highly dependent on characteristics associated with crystal nature and quality and that they are not useful as absolute values [50].
18.3.6 Comparison of Force Constants with Forces Measured by AFM
The force constants underlying thermal motions (20–200 pN/˚A) are of the same order of magnitude as forces explored by atomic force microscopy (AFM) on PM samples [51, 52]. The surface structure of BR has been explored by AFM and an applied force of 200 pN was necessary to bend away the E–F loop by 2 ˚A [53]. E ective force constants of thermal fluctuations represent an average value of the entire protein or the labeled part and are certainly higher than what can be expected for the flexible loop regions.
BR helices have been pulled o the membrane in pairs by applying forces around 350 pN [52]. The ruptured bonds correspond to interhelix and intrahelix hydrogen bonding and interaction with lipids and are, therefore, considerably higher than the bending forces.
18 Relating Protein Dynamics to Function and Structure |
431 |
To pull o lipids, a force of only 25 pN is su cient, as the interactions are smaller due to the fluid-like lipid behavior. This suggests that the force constants derived from thermal motions of lipids should be much smaller than those of the protein. Indeed, quasielastic incoherent neutron scattering on lipid bilayers have characterized fast in-plane and out-of-plane motions of lipid chains and molecules with amplitudes rising up to 3 ˚A [54].
18.4 Protein Dynamics and Function
in Some Other Proteins
As outlined in the preceding sections, biological activity of BR (i.e., proton pumping) is temperature-dependent. In particular, the ability of BR to complete its photocycle is abolished below the dynamical transition [24]. A correlation between the onset of biological function and the dynamical transition has been established in other proteins as well. In myoglobin, e.g., escape of the CO molecule from the heme pocket after flash photolysis is only possible at temperatures above the dynamical transition determined by M¨ossbauer spectroscopy [55]. Evidence for a dynamical transition at 220 K in Ribonuclease A has been obtained from the temperature-dependence of atomic B-factors [56]. Substrate-binding to Ribonuclease A is possible above, yet not below 220 K as revealed by temperature-controlled protein crystallography [57], thus suggesting a link between the dynamical transition and enzyme activity. Protochlorophyllide oxidoreductase is a light-activated enzyme and intermediates in the reaction pathway can be monitored by temperature-dependent absorption and fluorescence spectroscopy after initiating catalysis at cryo-temperatures (e.g., 100 K, [58]). The final step in the reaction pathway can only proceed above 200 K [59], a temperature close to the dynamical transition of many proteins. Despite these examples, a possible link between the dynamical transition and onset of biological activity remains a controversially discussed issue. Daniel and collaborators [60] conducted parallel measurements of dynamics and activity of glutamate dehydrogenase in a cryo-solution. Incoherent elastic neutron scattering experiments in the temperature range from 80 to 300 K revealed a dynamical transition of motions faster than 100 ps at 220 K. Enzymatic activity of glutamate dehydrogenase in the same cryo-solvent did not deviate from Arrhenius behavior in the measured temperature range from 280 down to 190 K. The authors concluded that the observed dynamical transition is decoupled from the rate-limiting step in the reaction pathway. However, the dynamical transition observed at 140 K for slower motions (i.e., faster than 5 ns) might, or might not be coupled to enzymatic activity, which cannot be measured below 190 K because of cryo-solvent freezing [61]. Not only fast motions probed by M¨ossbauer and neutron spectroscopy, but also much slower protein motions ( s–ms) measured by NMR have been identified as being involved in enzyme catalysis [62,63]. Even though protein dynamics is generally acknowledged to be crucial for activity, it remains unclear for proteins less
432 U. Lehnert et al.
well-studied than BR, in how far motions on di erent timescales are coupled to each other and which ones are essential for biological activity.
18.5 Conclusions
A general picture of the relation between thermal motions, structure and function emerges from the study of PM samples. The thermal motions reflect the protein flexibility that is necessary to access di erent conformational substates, but do not represent the conformational change in itself for which additional light energy is required to cross the barrier. The energy that is necessary for large conformational changes to occur during the photocycle has been provided by the light absorption and the thermal relaxations lead to the di erent photointermediates. Thermal motions help to overcome barriers between the di erent intermediates, but they do not directly reflect the conformation of the protein. Thermal fluctuations have been described as “. . . searching out the path or paths along which the transition takes place.” [4]. They are needed to prepare a “soft” environment in which the large conformational changes can take place and where the photointermediates are maintained in a stable configuration.
Acknowledgments
We thank G. Zaccai and F. Gabel for many fruitful discussions and for critical reading of the manuscript. U. Lehnert thanks the German Academic Exchange Service (DAAD) for a Postdoctoral fellowship. Support by the European Union under contract numbers HPRI-CT-2001-50035 and RII3-CT- 2003-505925 is gratefully acknowledged.
References
1.J.A. McCammon, S.C. Harvey, Dynamics of Proteins and Nucleic Acids (Cambridge University Press, Cambridge, 1987)
2.J.C. Smith, Quat. Rev. Biophys. 24, 227–291 (1991)
3.F. Gabel, D. Bicout, U. Lehnert, M. Tehei, M. Weik, G. Zaccai, Q. Rev. Biophys. 35, 327–367 (2002)
4.M. Karplus, J.A. McCammon, Ann. Rev. Biochem. 53, 263–300 (1983)
5.H. Frauenfelder, P.J. Steinbach, R.D. Young, Chem. Scripta 29A, 145–150 (1989)
6.H. Frauenfelder, S.G. Sligar, P.G. Wolynes, Science 254, 1598–1603 (1991)
7.G.R. Kneller, J.C. Smith, J. Mol. Biol. 242, 181–185 (1994)
8.G.R. Kneller, Chem. Phys. 261, 1–24 (2000)
9.K. Hinsen, A.-J. Petrescu, S. Dellerue, M.-C. Bellissent-Funel, G.R. Kneller, Chem. Phys. 261, 25–37 (2000)
18 Relating Protein Dynamics to Function and Structure |
433 |
10.J.A. Rupley, G. Careri, Adv. Protein Chem. 41, 37–172 (1991)
11.F. Parak, E.W. Knapp, D. Kucheida, J. Mol. Biol. 161, 177–194 (1982)
12.W. Doster, S. Cusack, W. Petry, Nature 337, 754–756 (1989)
13.L. Cordone, M. Ferrand, E. Vitrano, G. Zaccai, Biophys. J. 76, 1043–1047 (1999)
14.L. Cordone, P. Galajda, E. Vitrano, A. Gassmann, A. Ostermann, F. Parak, Eur. Biophys. J. 27, 173–176 (1998)
15.G. Cottone, L. Cordone, G. Ciccotti, Biophys J. 80, 931–938 (2001)
16.G. Zaccai, Science 288, 1604–1607 (2000)
17.D.J. Bicout, G. Zaccai, Biophys. J. 80, 1115–1123 (2001)
18.M.C. Bellissent-Funel, J.M. Zanotti, S.H. Chen, Faraday Discuss. 103, 281–294 (1996)
19.V. R´eat, H. Patzelt, M. Ferrand, C. Pfister, D. Oesterhelt, G. Zaccai, Proc. Natl. Acad. Sci. USA 95, 4970–4975 (1998)
20.A.E. Blaurock, J. Mol. Biol. 93, 139–158 (1975)
21.U. Haupts, J. Tittor, D. Oesterhelt, Annu. Rev. Biophys. Biomol. Struct. 28, 367–399 (1999)
22.J.K. Lanyi, B. Schobert, Biochemistry 43, 3–8 (2004)
23.G. Zaccai, J. Mol. Biol. 194, 569–572 (1987)
24.M. Ferrand, A.J. Dianoux, W. Petry, G. Zaccai, Proc. Natl. Acad. Sci. USA 90, 9668–9672 (1993)
25.U. Lehnert, V. R´eat, M. Weik, G. Zaccai, C. Pfister, Biophys. J. 75, 1945–1952 (1998)
26.R. Neutze, E. Pebay-Peyroula, K. Edman, A. Royant, J. Navarro, E.M. Landau, Biochim. Biophys. Acta 1565, 144–167 (2002)
27.N.A. Dencher, H.J. Sass, G. B¨uldt, Biochim. Biophys. Acta 1460, 192–203 (2000)
28.V. R´eat, G. Zaccai, M. Ferrand, C. Pfister, Functional dynamics in purple membrane, in Biological Macromolecular Dynamics (Adenine, Guilderland, Newyork, 1997) pp. 117–122
29.J. Fitter, R.E. Lechner, G. B¨uldt, N.A. Dencher, Proc. Natl. Acad. Sci. USA 93, 7600–7605 (1996)
30.J. Fitter, R.E. Lechner, N.A. Dencher, Biophys. J. 73, 2126–2137 (1997)
31.J. Fitter, R.E. Lechner, N.A. Dencher, Physica B 226, 61–65 (1996)
32.H.J. Sass, I.W. Schachowa, G. Rapp, M.H.J. Koch, D. Oesterhelt, N.A. Dencher,
G.B¨uldt, EMBO J. 16, 1484–1491 (1997)
33.M. Weik, G. Zaccai, N.A. Dencher, D. Oesterhelt, T. Hauss, J. Mol. Biol. 275, 625–634 (1998)
34.R.E. Lechner, J. Fitter, N.A. Dencher, T. Hauss, J. Mol. Biol. 277, 593–603 (1998)
35.M. Weik, U. Lehnert, G. Zaccai, Biophys. J. 29, 29 (2005)
36.U. Lehnert, V. R´eat, B. Kessler, D. Oesterhelt, G. Zaccai, Appl. Phys. A 74, S1287-S1289 (2002)
37.J. Fitter, S.A. Verclas, R.E. Lechner, H. Seelert, N.A. Dencher, FEBS Lett. 433, 321–325 (1998)
38.H. Belrhali, P. Nollert, A. Royant, C. Menzel, J.P. Rosenbusch, E.M. Landau,
E.Pebay-Peyroula, Struct. Fold Des. 7, 909–917 (1999)
39.P. Kraulis, J. Appl. Cryst. 24, 946–950 (1991)
40.E.A. Merritt, D.J. Bacon, Methods Enzymol. 277, 505–524 (1997)
41.J. Fitter, O.P. Ernst, T. Hauss, R.E. Lechner, K.P. Hofmann, N.A. Dencher, Eur. Biophys. J. 27, 638–645 (1998)
434 U. Lehnert et al.
42.C.L. Brooks, M. Karplus, B.M. Pettitt, Proteins: a theoretical perspective of dynamics, structure, and thermodynamics, in Advances in Chemical Physics,
I.Rice, S.A. Prigogine (Eds.) (John Wiley & Sons, New York, 1988) pp. 94–95.
43.G. V´ar´o, J.K. Lanyi, Biochemistry 30, 5008–5015 (1991)
44.J. Le Coutre, L.R. Narasimhan, C.K. Patel, H.R. Kaback, Proc. Natl. Acad. Sci. USA 94, 10167–10171 (1997)
45.J. Abramson, I. Smirnova, V. Kasho, G. Verner, H.R. Kaback, S. Iwata, Science 301, 610–615 (2003)
46.N. Grigorie , T.A. Ceska, K.H. Downing, J.M. Baldwin, R. Henderson, J. Mol. Biol. 259, 393–421 (1996)
47.O. Edholm, O. Berger, F. J¨ahnig, J. Mol. Biol. 250, 94–111 (1995)
48.H. L¨ucke, B. Schobert, H.T. Richter, J.P. Cartailler, J.K. Lanyi, J. Mol. Biol. 291, 899–911 (1999)
49.H.J. Sass, G. B¨uldt, R. Gessenich, D. Hehn, D. Ne , R. Schlesinger, J. Berendzen, P. Ormos, Nature 406, 649–653 (2000)
50.J.B. Heymann, D.J. M¨uller, E.M. Landau, J.P. Rosenbusch, E. Pebay-Peyroula,
G.B¨uldt, A. Engel, J. Struct. Biol. 128, 243–249 (1999)
51.D.J. M¨uller, G. B¨uldt, A. Engel, J. Mol. Biol. 249, 239–243 (1995)
52.F. Oesterhelt, D. Oesterhelt, M. Pfei er, A. Engel, H.E. Gaub, D.J. M¨uller, Science 288, 143–146 (2000)
53.D.J. M¨uller, H.-J. Sass, S.A. M¨uller, G. B¨uldt, J. Mol. Biol. 285, 1903–1909 (1999)
54.S. K¨onig, W. Pfei er, T. Bayerl, D. Richter, E. Sackmann, J. Phys. II France 2 8, 1589–1615 (1992)
55.H. Lichtenegger, W. Doster, T. Kleinert, A. Birk, B. Sepiol, G. Vogl, Biophys.
J.76, 414–422 (1999)
56.R.F. Tilton, J.C. Dewan, G.A. Petsko, Biochemistry 31, 2469–2481 (1992)
57.B.F. Rasmussen, A.M. Stock, D. Ringe, G.A. Petsko, Nature 357, 423–424 (1992)
58.D.J. Heyes, A.V. Ruban, H.M. Wilks, C.N. Hunter, Proc. Natl. Acad. Sci. USA 99, 11145–11150 (2002)
59.D.J. Heyes, A.V. Ruban, C.N. Hunter, Biochemistry 42, 523–528 (2003)
60.R.M. Daniel, J.C. Smith, M. Ferrand, S. Hery, R. Dunn, J.L. Finney, Biophys.
J.75, 2504–2507 (1998)
61.R.M. Daniel, J.L. Finney, V. R´eat, R. Dunn, M. Ferrand, J.C. Smith, R.V. Dunn, J. Finney, Biophys. J. 77, 2184–2190 (1999)
62.F.A. Mulder, A. Mittermaier, B. Hon, F.W. Dahlquist, L.E. Kay, Nat. Struct. Biol. 8, 932–935 (2001)
63.E.Z. Eisenmesser, D.A. Bosco, M. Akke, D. Kern, Science 295, 1520–1523 (2002)

19
Biomolecular Spectroscopy
Using Pulsed-Source Instruments
H.D. Middendorf
19.1 Introduction
Neutron scattering techniques have already been applied to a wide range of problems in biomolecular dynamics, from anhydrous building blocks to a variety of partially or fully hydrated biomolecules and their assemblies [1–7]. However, much of what has been published so far deals with data sets that are very limited in relation to the structural and dynamical complexities of the systems studied, and the impact of this work in the life sciences has been small.
There is a broad concensus that increases in data collection rates by 1–2 orders of magnitude are required if the full potential of neutron scattering as a more widely used tool is to be realized [8–10]. Only high-intensity pulsed sources can deliver increases of this magnitude. A factor of 10–20 will be needed to bring sample sizes down into the 20–50 mg region. The 1/2 g quantities required for present-day neutron spectroscopy are much too large. Another factor of 10 could be “spent” either on higher-quality data sets from experiments designed to reveal functionally significant di erence spectra, or on more extensive coverage of the large parameter domain of biomolecular systems.
19.2 Why Pulsed Sources?
It has been recognized since the 1980s [11,12] that there is little room for flux increases from fission reactors beyond the level achieved by the world’s most powerful steady-state neutron source at the Institut Laue-Langevin (ILL) in Grenoble. In Europe, the only new research reactor built during the past 20 years (FRM-II at Munich) requires highly enriched uranium for optimal performance, a politically contentious design option that is unlikely to be adopted ever again.
436 H.D. Middendorf
SPALLATION
GeV proton
FISSION
eV neutron
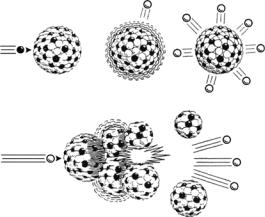
Fig. 19.1. A GeV proton incident on a heavy-metal nucleus leads to an excited state that emits 15–25 spallation neutrons, but the nucleus does not undergo fission. In the fission case, a uranium nucleus produces on average only 2.5 neutrons, one of which is needed to sustain the chain reaction
An alternative way of producing intense neutron beams is by bombarding a block of heavy metal with GeV proton pulses [11]. Each incident proton “spalls” between 15 and 25 neutrons o a target nucleus (Fig. 19.1). In addition to cold and thermal neutron beams from appropriate moderators, spallation sources provide epithermal neutrons, which open up possibilities for electron volt spectroscopy and medical applications. Following pioneering work at research centers with low-intensity accelerators, the technology of such sources was advanced significantly by the commissioning in 1986 of the first high-intensity spallation source, the ISIS facility at the Rutherford Appleton Laboratory near Oxford, England. New facilities are under construction currently at Tokai (JSNS [13]), Japan, and at Oak Ridge (SNS [14]) USA, and a second target station is being built at ISIS (TS2 [15]).
Pulsed-source instruments can achieve large gain factors even with inferior time-averaged fluxes. By cutting out a series of spikes from a continuous white beam and monochromatising them, conventional choppers always “throw away”’ more than 99.9% of incident neutrons. With a pulsed beam, on the other hand, neutrons already arrive “concentrated” as spikes separated by 20–100 ms according to the duty cycle of the accelerator. Since the source is e ectively “switched o ” between successive pulses, scattered neutrons can be energy-analyzed with excellent signal/noise ratios. A drawback of fixed-frequency pulsed sources is that the gain factors of instruments covering di erent energy regions cannot be optimized simultaneously. By constructing a second target station running at a lower frequency, ISIS is the only facility aiming for the best of both worlds.

|
|
19 |
Biomolecular Spectroscopy |
437 |
|||
|
|
Table 19.1. Spallation neutron sources |
|
|
|
||
|
|
|
|
|
|
|
|
Country |
Name |
Institution |
Location |
Current |
Frequency |
||
|
|
|
|
( A) |
(Hz) |
|
|
UK |
ISISa |
Rutherford Appleton Lab |
Chilton |
200 |
50/10 |
|
|
CH |
SINQb |
Paul Scherrer Institut |
Villigen |
1,300 |
qu.cont. |
|
|
USA |
IPNS |
Argonne National Lab. |
Argonne |
15 |
30 |
|
|
USA |
LANSCE Los Alamos National Lab |
Los |
100 |
20 |
|
|
|
|
|
|
Alamos |
|
|
|
|
USA |
SNSa |
Oak Ridge National Lab |
Oak |
1,300 |
60 |
|
|
|
|
|
Ridge |
|
|
|
|
Japan |
KENS |
KEK |
Tsukuba |
6 |
20 |
|
|
Japan |
JSNSa |
J-PARC (KEK-JAERI) |
Tokai |
330 |
25 |
|
|
aunder construction: SNS, JSNS, ISIS-TS2 (10-Hz target station of ISIS) bquasi-continuous (50 MHz)
At present there are five neutron research centers operating spallation sources, and four of these are pulsed sources (Table 19.1). Instruments at SINQ (Paul Scherrer Institute, Switzerland) are not able to exploit the inherent time structure of a pulsed beam since the source is quasi-continuous. In the following the discussion will be limited to instruments and experiments at the four spallation sources producing pulsed beams.
19.3 Pulsed Source vs. Reactor Instruments
Neutron scattering experiments using pulsed-source instruments are not in principle di erent from those on reactor-source instruments. With the exception of the spin-echo technique [16, 17], the objective in both is to measure double-di erential cross-sections d2σ/dΩ dE from which dynamic structure factors Sinc(Q, ω) and Scoh(Q, ω) for incoherent and coherent scattering can be extracted [11] ( Q = momentum transfer, ω = energy transfer). However, there are significant di erences in instrument design and operation that may a ect experimental strategy, and there are also instrument types covering Q, ω-regions that are not or only partly accessible by spectrometers on conventional sources. Most applied-science users familiar with instruments on the latter will appreciate the larger dynamic range and (for backscattering) better Q-resolution of pulsed-source spectrometers.
A comparison of the Q, ω-envelopes of spectrometers at ILL and ISIS (Fig. 19.2) shows an area of overlap flanked by regions of lower and higher energy transfers, which at present are accessible only by reactor-source (lowω) or pulsed-source instruments (high ω). While this reflects the fact that the energy distribution of raw (i.e., undermoderated) spallation neutrons is centered at higher energies than that of fission neutrons, the complementarity