

Neutron Scattering in Biology - Fitter Gutberlet and Katsaras
.pdf21 Internal Dynamics of Proteins and DNA |
489 |
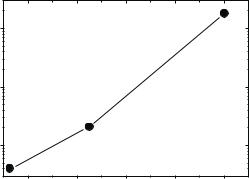
(Fig. 21.3). The stretching exponents β (stretching in the time domain) and b (stretching in the frequency domain) are not identical, but they are directly related [13]. The value of the exponent b is smaller than in most of glassforming systems, i.e., the stretching of the slow relaxation process in proteins and DNA is stronger than in molecular liquids or polymers, suggesting a more complex relaxation process or a broader distribution of relaxation times. The contribution of the slow process to the susceptibility spectrum changes strongly with temperature (Fig. 21.3). The changes of the spectra are usually ascribed to temperature variations of the characteristic relaxation time τS(T ). Analysis of the spectra (Fig. 21.3a) reveals a strong temperature dependence of τS(T ) in DNA at T > 250 K (Fig. 21.4) such that the slow process moves out of the accessible frequency range at T < 210–230 K and makes estimates of τS unreliable. A strong (usually nonArrhenius) temperature dependence of the relaxation time and stretching of the relaxation spectrum are characteristic of structural relaxation in glass-forming liquids. Thus the slow process in
|
103 |
320K |
|
|
|
|
|
|
|
|
|
290K |
|
|
|
|
250K |
|
|
n)(arb.un.) |
102 |
|
210 K |
(a) |
|
|
|||
|
320K |
|
|
|
c"( |
103 |
290K |
|
|
|
|
|
|
|
|
|
320K |
|
|
|
102 |
150K |
|
|
|
|
|
|
|
|
|
150K |
|
(b) |
|
|
|
|
|
|
100 |
101 |
102 |
103 |
|
|
n (GHz) |
|
|
Fig. 21.3. Temperature variations of susceptibility spectra for DNA at various hydration levels: (a) 0.46 g of D2O per gram of DNA, (b) 0.66 g of D2O per gram of DNA (open symbols) and 0.03 g of D2O per gram of DNA (closed symbols) (for details, see [3]). Thin solid lines show the fit to Eq. 21.4. The thick solid line shows the spectrum of the fast process χfast(ν)
490 A.P. Sokolov et al.
|
1 10−8 |
|
|
|
|
|
(s) |
1 10 |
−9 |
|
|
|
|
s |
|
|
|
|
|
|
t |
|
|
|
|
|
|
1 10−10 |
|
|
|
|
||
|
|
3.2 |
3.4 |
3.6 |
3.8 |
4.0 |
|
|
|
|
1000/T (1/K) |
|
|
Fig. 21.4. Temperature dependence of the characteristic relaxation time τS of the slow relaxation process in hydrated DNA (Fig. 21.3a) obtained from a fit to Eq. 21.4.
biological macromolecules appears to be similar to the main structural relaxation process in glass-forming liquids [14–16].
There is, however, one significant di erence between the structural relaxation (α-relaxation) in glass-forming liquids and the slow process in biological macromolecules. It appears in the Q-dependence of the characteristic relaxation time. The structural relaxation time, τα in glass-forming systems has a strong dependence on Q, i.e., τα Q−c, with c 2–4 [17, 18] indicative of di usive motion. For particles undergoing free translational di usion, the van Hove correlation function obeys Fickian di usion and the correspond-
ing |
intermediate scattering function is given by I(Q, t) = exp( |
t/τ |
α |
) with |
||
|
2 |
− |
|
|
||
τα |
1/DQ |
where D is the di usion coe cient. By contrast, analysis of |
||||
τS in proteins reveals no significant Q-dependence [1, 4], consistent with the slow relaxation process in proteins involving localized di usive motions with atomic displacements smaller than 3 ˚A. If di usion is confined to a sphere of radius r, the relaxation time is independent of Q for Q < π/r and varies as 1/DQ2 for Q values > π/r [19]. Using a two-site jump model, Doster and Settles [16] estimated the jump distance to be 1.5 ˚A. Such confined di usive motion of groups of atoms in the interior of proteins is to be expected given the compact folding of the polypeptide backbone in the native state.
The microscopic picture behind the slow process remains unclear. However, results of recent computer simulations [20] provide additional insight. The authors found that a few collective modes distributed over the whole protein molecule are activated above T 180–220 K. These modes represent small displacements of secondary structures (α-helices) of the protein. Because the slow process is also activated at this temperature range, we tend to ascribe it to these kinds of modes. That will explain weak Q-dependence of the relaxation time.
21 Internal Dynamics of Proteins and DNA |
491 |
There is much evidence for water acting as a plasticizer of proteins including ESR studies of spin labels [21–23], M¨ossbauer spectroscopy [22, 24], phosphorescence [25,26], solid-state 13C cross polarization/magic angle sample spinning (CP/MASS) NMR [27], hydrogen isotope exchange [28,29], dielectric relaxation [30, 31], Rayleigh scattering of M¨ossbauer radiation (RSMR) [32], light-scattering [12] and incoherent neutron scattering [1, 3, 4, 33]. The main conclusion to be draw from these studies is that at 300 K the internal dynamics of proteins recovers at hydration levels of 0.1–0.2 g water g protein−1, much lower than the amounts of water required for full hydration (0.4–0.5 g water g protein−1). In fully hydrated systems, internal dynamics recover at temperatures of about 180–220 K.
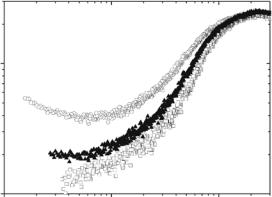
The detailed mechanism by which water plasticizes proteins and the coupling of the solvent to the protein interior are not fully understood. It has been suggested that water plasticizes proteins in the same way it plasticizes polyamides such as Nylon 6,6 and Nylon 4,6 by providing alternate, mobile hydrogen bond donors and acceptors for peptide groups [34]. This may involve the migration of buried water molecules in the protein interior via “Mobile Defects” in the hydrogen bonding network [35] or could be due to water at the protein surface. Several recent simulations [36–38] suggest that the dynamics of water at the protein surface activates protein internal dynamics. Recent neutron scattering studies of the e ect of hydration on proteins and DNA clearly show that the slow relaxation process occurs in the hydrated systems but not in the dry systems (Figs. 21.2, and 21.3b) [3, 4, 12], suggesting that water enables the slow relaxation process.
There is also evidence that the slow relaxation process occurs in proteins in glycerol (Fig. 21.5). The contribution of the slow process is weaker in the case of lysozyme in glycerol and nearly disappears from the accessible frequency range at T 250 K. There has been much discussion of the e ects of cosolvents such as glycerol and sugars on proteins. The relationship between the solvent viscosity and kinetics of biochemical reactions has been discussed in many papers [39–46]. Increases in solvent viscosity can increase internal friction associated with molecular motions and slow down protein dynamics. But cosolvents also have e ects on the thermodynamics of proteins. It is wellestablished that polyols such as glycerol and sugars increase protein chemical potentials. As the protein–water–polyol system adjusts to relieve the unfavorable increase in free energy, the polyols are preferentially excluded from the vicinity of the protein (the protein is preferentially hydrated) [47] and the protein favors more compact conformations in order to minimize its contact with the polyol. The later e ect may have a large influence on protein internal dynamics as has been suggested for the e ect of glycerol on protein hydrogen isotope exchange [48].
We should emphasize that the appearance of the slow relaxation process in the presence of glycerol, shown in Fig. 21.5, occurs in the absence of water. These neutron scattering results in glycerol [4] are also supported by parallel light scattering measurements [12] and clearly indicate that glycerol itself can

21 Internal Dynamics of Proteins and DNA |
493 |
|
|
Wet lysozyme |
|
|
|
|
|
0.15 |
Lysozyme/Glycerol |
|
|
|
|
|
|
Lysozyme/Trehalose |
|
|
|
|
) |
|
|
|
|
|
|
2 |
0.10 |
|
|
|
|
|
>(A |
|
|
|
|
|
|
|
|
|
|
|
|
|
2 |
|
|
|
|
|
|
<x |
|
|
|
|
|
|
|
0.05 |
|
|
|
|
|
|
0.00 |
|
|
|
|
|
|
50 |
100 |
150 |
200 |
250 |
300 |
T(K)
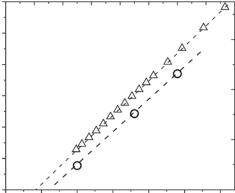
Fig. 21.6. Mean-squared displacements of atoms <x2> in lysozyme dissolved in various solvents (data from [4]). The dynamical transitions for hydrated lysozyme appears at TD 220 K while it appears in lysozyme embedded in glycerol at TD 270 K. No dynamic transition appears in lysozyme embedded in trehalose up to T 320 K
does not appear in dry proteins and DNA even at temperatures as high as T 320 K [11, 51, 52]. Moreover, the transition can be strongly influenced by solvent (Fig. 21.6), shifting to higher T 270–280 K for proteins embedded in glycerol [4, 51] and being suppressed at least up to T 320 K in proteins formulated in solid trehalose [4,54]. All these results suggest that solvents play a crucial role in the dynamical transition and activity of proteins.
The mean square displacement, <x2>, is an integrated quantity that usually includes contributions from vibrations, conformational changes, diffusion and other molecular motions. Analysis of time or frequency resolved spectra might therefore help to understand which processes are activated as the temperature is increased above TD. Neutron spectroscopy is an extremely useful tool for these measurements because it provides information over a wide range of frequencies. These kinds of measurements have been performed on only a few proteins and on DNA [3, 7, 9, 12] and indicate that the dynamical transition is associated with activation of the slow relaxation process that enters the Gigahertz frequency window at temperatures T > TD 200–230 K (Fig. 21.3).
The microscopic mechanism of the dynamical transition remains a subject of much discussion. Mode coupling theory (MCT) developed in the mideighties [55] to describe the dynamics of simple liquids, supercooled liquids and glass-forming systems may provide a useful framework for understanding the dynamic behavior of proteins and DNA. MCT predicts a dynamic crossover in glass-forming liquids marking a transition from liquid-like to solid-like behavior on molecular time and length scales at temperatures much above conventional glass transition temperatures. Many experimental results support the
494 A.P. Sokolov et al.
MCT predictions [56], and the theory gives a reasonably good qualitative description of relaxation spectra in many glass-forming systems at temperatures above the crossover temperature TC, although other experimental observations are not in accord with idealized MCT [57].
MCT predicts that relaxation in glass-forming systems at high temperatures (T > TC) occurs by a two-step process involving an initial fast decay corresponding to relaxation of a particle (a molecular unit) confined (“rattling”) in a cage formed by its neighbors; which decays to a particular level but does not lead to a complete relaxation; followed by a slow second step that corresponds to escape of the particle from the cage. The theory predicts that both relaxation processes are described by stretched decays. Details of MCT predictions and its experimental tests can be found in various reviews (see, for example [55, 56]).
On a qualitative level, MCT predicts that at T > TC the fast process is essentially temperature independent and the main variation is a strong slowing down of the slow relaxation process [55]. Moreover, the characteristic relaxation time of the slow relaxation process should exhibit a critical temperature
dependence |
|
τslow(T ) (T − TC)−γ |
(21.2) |
where the value of the critical exponent γ is directly related to the stretching exponent b [55]. This provides a reasonably good qualitative description of data from various ionic and van der Waals molecular systems, polymers, as well as covalent and hydrogen bonded liquids [56, 58, 59], including water and glycerol [60, 61].
Doster et al. [7] applied MCT to describe neutron scattering spectra for myoglobin. They showed that the theory can describe the spectra and their temperature variations, and estimated a crossover temperature TC 195 K. Sokolov et al. performed an analysis of neutron scattering spectra of DNA at various levels of hydration [3, 9] and demonstrated (Fig. 21.3) that MCT describes well the qualitative features of the dynamics of DNA down to temperatures of about 230 K. The authors fit the susceptibility spectra by a sum of a slow and a fast relaxation process
χ (ν, T ) = χslow(ν, T ) + χfast(ν) = A[ντS(T )]b + χfast(ν). |
(21.3) |
Here the slow process was approximated by a power law with a stretching exponent b 0.3. All parameters, except τS(T ) were assumed to be temperature independent and τS(T ) was the only free fitting parameter [3]. Its temperature dependence is presented in Fig. 21.4. This high-temperature MCT description works well down to T 230 K. Below this temperature, Eq. 21.4 cannot provide a good description of the spectra because the fast process becomes temperature dependent [3, 9]. This corresponds to the dynamic crossover and gives an estimate of TC 230 K [3, 9]. As noted earlier, MCT also predicts temperature variations of τS(T ) (Eq. 21.3), so the temperature dependence of the only free fitting parameter can be verified. The exponent γ in Eq. 21.3 is related to the
496 A.P. Sokolov et al.
the correlation between TD of a protein and TC of a solvent? Results of several recent computer simulations provide some insight. In one case, the di usion of water molecules was constrained by placing the oxygen atoms in a very deep energy minimum. The water molecules could rotate, and the lifetime of hydrogen bonds remains similar to the unrestricted case, however, analysis of the intermediate scattering function I(Q, t) of the protein in an environment with restricted water di usion was found to resemble I(Q, t) for the dry protein [36]. The slow process appears to be suppressed and restriction of water di usion places the hydrated protein below its dynamic transition even at room temperature [36].
A recent molecular dynamics simulation employing a dual heat bath method, in which the temperature of the solvent and protein could be maintained at di erent values during the simulation, shows that the protein dynamical transition is driven by a dynamical transition in the translational motion of water [37]. The latter enables increased fluctuations of the protein especially in the motions of side chains at the protein surface. According to MCT, the dynamic crossover is essentially a dynamic arrest on a molecular time and length scale. In this case, decreasing the temperature below TC of a solvent is equivalent to restricting translational di usion of the solvent molecules. This restriction brings the protein below its dynamical transition. This picture provides a microscopic explanation for the observed correspondence of the solvent dynamic crossover temperature TC and the protein dynamical transition temperature TD. It agrees with the idea proposed some time ago [43] that proteins are “slaves” of the solvent. The dynamics of water bonded to the protein surface are known to di er from the dynamics of bulk water, which might a ect the TC of solvent molecules at the protein interface, but this e ect might be of secondary importance.
21.5 Fast Picosecond Relaxation
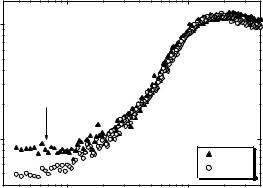
In this section we focus our attention on the fast relaxation process. It is present in the spectra of all proteins even at very low temperatures. It overlaps with the boson peak vibrations and has a stretched low-frequency tail (Fig. 21.2). Changes in temperature lead to a change in amplitude rather than a change in relaxation time (Fig. 21.3b). All these properties are known for the fast relaxation process in glass-forming systems [63]. Nevertheless, the microscopic picture of the fast relaxation in proteins and DNA remains unclear. The small mean square atomic displacements <x2> associated with the fast relaxation process (see for example, the data for <x2> shown in Fig. 21.6) suggest it is a local process, and we interpret it as involving fast conformational fluctuations of side-chains, and perhaps peptide groups. These fast conformational fluctuations might be the necessary precursors for global conformational transitions in proteins that happen at much longer times.
498 A.P. Sokolov et al.
or base-pairs. Fast fluctuations of peptide groups may also contribute to the fast relaxation process. 2H NMR studies of spin–lattice relaxation times of 2H-labeled lysozyme provide evidence of small amplitude (9◦–12◦) torsional motions with correlation times of about 15 ps [66].
The energy landscape approach is similar to the idea of hierarchical states proposed for proteins by Frauenfelder and coworkers [45]. Using this approach, one can estimate the distribution of barrier heights g(V ) between substates. This approach has been used for analysis of susceptibility spectra of some glass formers [63, 67]. In a simple approximation one can estimate g(V ) from the measured χ (ν) [63, 67]
g(V ) χ (ν)/T ; where V = −T ln(2πντ0). |
(21.4) |
The nature of the fast relaxation can be analyzed using coherent neutron scattering which can provide microscopic information on the molecular motions involved, and the cooperativity of motions of amino-acid residues.
The role of the fast relaxation in the function of biological molecules is not understood. Fast fluctuations are thought to be a necessary precursor for any slower processes, including those important for protein function. Suppression of the fast relaxation process in proteins by putting them in various sugar formulations is known to greatly increase their deactivation time [68, 69].
21.6 Conclusions and Future Prospects
The dynamics of proteins and nucleic acids have been the subject of intense study with a variety of experimental methods and computational modeling. Neutron spectroscopy occupies a special position in such studies (along with techniques such as Rayleigh Scattering of M¨ossbauer radiation) because the experimental observable, the double di erential scattering cross-section related to the dynamic structure factor, combines timeand space-correlations in molecular motion. The combination of neutron spectroscopy with computer simulations, in our view, is emerging as one of the best approaches to study the dynamics of complex systems such as proteins. This combination helps to verify parameters of computer modeling and to visualize the motions that appear in scattering functions probed by neutrons.
One very promising direction is the analysis of the dynamics of selectively deuterated proteins [70, 71]. Analysis of selectively deuterated synthetic polymers is a well-developed method that provides rather precise information on the motion of particular parts of the molecule. It is now possible to synthesize proteins with cell-free protein synthesis systems that will allow specific deuterated amino acids to be incorporated into the protein or for all aminoacid residues in the protein to be deuterated. The main problem is in the quantities of sample required for modern neutron scattering spectroscopy – usually 100 mg or more. Production of this amount of deuterated protein