

Molecular and Cellular Signaling - Martin Beckerman
.pdf16.3 TFs Bind DNA Through Their DNA-Binding Domains 391
FIGURE 16.4. CREB bZIP dimer bound to DNA determined using X-ray crystallography: The basic region (residues 285 to 307 straddle and grip the DNA helix while leucine zipper residues 308 through 336 form a dimerization interface. The basic residues contact the major groove of the CRE region in the promoter. Protein helices are depicted as ribbons and DNA molecules as balls-and-sticks. The figure was prepared using Protein Explorer with atomic coordinates deposited in the Protein Date Bank under accession number 1DH3.
are interdigitized with the leucines from the partner molecule to form a short coiled-coil dimerization region, hence the name “leucine zipper.” These proteins also contain a region rich in basic residues that interfaces with the DNA. The basic region begins at a location seven residues N- terminal to the first leucine in the leucine zipper domain. The overall domain is referred to as a basic region leucine zipper (bZIP) domain; the resulting bZIP dimer tightly grips the DNA molecule in the major groove as illustrated in Figure 16.4 for the CREB transcription factor (CREB transcription factors will be discussed in Chapter 21). A number of transcription factors possess a basic region plus HLH and leucine zipper domains. These DNA-binding proteins are referred to as bHLH-LZ proteins. The bHLH-LZ and bZIP proteins form a variety of homeodimers and heterodimers with one another.
The final category of DNA-binding proteins to be discussed is the zinc fingers. These proteins utilize one or two zinc atoms to help form compact domains of about 30 amino acid residues. The zinc finger motif is often repeated, and each small module is organized about a central zinc atom.
Adjacent zinc finger modules form an overall DNA-binding domain whose zinc fingers grip the DNA molecule. This motif is widely distributed among eukaryotic transcription factors. The specifc composition of these modules is variable but all contain zinc atoms and independently fold into small compact structures as depicted in Figure 16.5.

392 16. Gene Regulation in Eukaryotes
FIGURE 16.5. Zinc fingers of the TFIIA protein in contact with DNA determined using X-ray crystallography: The DNA-binding domain of the transcription factor TFIIA has six zinc fingers each organized by a central zinc atom (indicated in the figure by small filled circles). Protein helices are depicted as ribbons and DNA molecules as balls-and-sticks. The figure was prepared using Protein Explorer with atomic coordinates deposited in the Protein Data Bank under accession number 1TF6.
16.4Transcriptional Activation Domains Initiate Transcription
The tight packaging of the DNA into nucleosomes inhibits transcription. Transcription factors stimulate transcription by: recruiting to transcription sites components of the transcription machinery; recruiting cofactors such as the Mediator; and by attracting to these sites molecules and complexes that remodel and modify chromatin structure.
Transcription factors possessing DNA-binding and dimerization domains alone cannot trigger transcription. Instead, an activation domain must be present. Transcription factors utilize several kinds of activation domains. These domains are enriched in specific amino acids and are classified according to their composition rather than by any particular secondary or supersecondary structures. Some are enriched in acidic amino acid residues and are called acid blobs. Others are enriched in glutamine or proline residues.
The activation domains of transcription factors are protein-binding regions. One of the ways the activation regions stimulate transcription is by recruiting components of the general transcriptional machinery. The transcription activators bind to sites along the DNA several hundred base units upstream of the transcription start site using their DNA-binding domains. Components of the transcription machinery are then recruited to and bind the activation domains, thus seeding the further assembly of transcription components and, so, stimulating onset of transcription.
Chromatin remodeling complexes are attracted to the promoter site by the activation domains of the transcription factors. Referring back to the yeast modules listed in Table 16.1, The SWI/SNF module is recruited by the acidic activation domain present in many transcription factors. The SAGA

16.6 Composition and Structure of the Basal Transcription Machinery |
393 |
|||
|
TABLE 16.2. Nuclear receptors and their ligands: Com- |
|
||
|
mon abbreviations for the receptors are given within the |
|
||
|
parentheses. |
|
|
|
|
Steroid receptors |
Nonsteroid receptors |
|
|
|
|
|
|
|
|
Androgens (AR) |
Ecdysone (EcR) |
|
|
|
Estrogens (ER) |
All-trans retinoids (RAR) |
|
|
|
Cortisol (GR) |
9-cis retinoids (RXR) |
|
|
|
Aldosterone (MR) |
Thyroid hormone (TR) |
|
|
|
Progestins (PR) |
Vitamin D3 (VDR) |
|
|
|
|
|
|
|
histone acetyltransferase module can also be recruited in this manner. The recruitment of the chromatin remodeling machinery is not dependent on the presence of either the TBP (TATA binding protein) or RNA polymerase II at the promoter.
16.5Nuclear Hormone Receptors Are Transcription Factors
The genomes of complex metazoans contain large numbers of transcription factors. An early estimate is that there are over 3000 transcription factors encoded in the human genome. Nuclear hormone receptors, with over 150 members, form the largest family of transcription factors. These receptors can be grouped into two large families, designated as Class I (steroid) and Class II (nonsteroid). Representative members of these two families are listed in Table 16.2.
Steroid hormones and other small lipophilic molecules such as vitamin
D3 are able to pass through the plasma membrane and enter the cell interior where they bind to nuclear receptors. Following ligand binding, the steroid receptors dimerize, translocate into the nucleus from the cytoplasm, and bind their cognate responsive elements. Class II receptors differ from those of Class I in that they can bind to responsive elements in the absence of ligand binding. Many Class II receptors readily form heterodimers and bind to a variety of coactivators.
16.6Composition and Structure of the Basal Transcription Machinery
The basal transcription machinery consists of RNA polymerase II and a set of general transcription factors (GTFs). These components, listed in Table 16.3, make up a minimal set of transcription initiation factors that support recognition and low (basal) levels of transcription from most promoter

394 16. Gene Regulation in Eukaryotes
TABLE 16.3. Basal transcription machinery: Listed are the components, the number of subunits in each component, their mass, and their role in initiating transcription (discussed further in the main text).
Component |
Number of subunits |
Molecular weight (kDa) |
Function |
TFIID-TBP |
1 |
38 |
TATA binding protein |
TFIID-TAFIIs |
12+ |
15–250 |
Positive/negitive regulators |
TFIIB |
1 |
35 |
TFIIF/Poly II binding |
TFIIF |
2 |
30, 74 |
Shuttles Poly II |
RNA Poly II |
12 |
10–220 |
Catalytic |
TFIIE |
2 |
34, 57 |
Stim. TFIIH melting |
TFIIH |
9 |
35–89 |
Stim. melting/escape |
|
|
|
|
sites. The minimal set of general transcription factors assembled at the promoter includes in order of recruitment TFIID, TFIIB, TFIIF, TFIIE, and TFIIH. Another GTF, TFIIA, is sometimes recruited to the promoter, and this (disposable) unit assists in TBP binding and stabilization. The assembly of the minimal set at the core promoter is depicted schematically in Figure 16.1(b).
TFIID is believed to be the first component of the transcription initiation complex to be recruited to the promoter site. It is the major sequencespecific GTF, and is a common target of TF activation domains. TFIID contains the TATA box-binding protein (TBP), and a collection of TBPassociated factors (TAFs). The TBP subunit is a general transcription factor. It recognizes the TATA sequence in its minor groove and is present in all RNA polymerization operations. The TAFs are promoter-selective and different mixes of these factors are present in different promoter sites. Recruitement of TFIID triggers the recruitment of TFIIA and then TFIIB followed by TFIIF bound to RNA polymerase II, and after that TFIIE, and then TFIIH. These additional GTFs that follow establish protein-protein contacts and link to the promoter site through the TBP. The TBP bends the DNA and forms a saddle with its inner surface in contact with the DNA molecule covering an 8-bp region of DNA.
16.7RNAP II Is Core Module of the Transcription Machinery
The RNAP II molecule is composed of 12 subunits designated as Rpb1 through Rpb12. The yeast RNAP II molecule contains 4565 amino acid residues and has a mass of 514 kDa. The sizes of the subunits vary considerably. The largest two, Rpb1 (192 kDa) and Rpb2 (139 kDa), account for the bulk of the mass of the polymerase. A top view of the entire assembly is presented in Figure 16.6.

16.8 Regulation by Chromatin-Modifying Enzymes |
395 |
FIGURE 16.6. Top view of RNAP II determined by means of X-ray diffraction measurements at 3.1 Å and 2.8 Å resolution: Ten of the subunits are shown in this figure. Superimposed on this structure is a representation of a DNA molecule entering the RNAP II, and an arrow indicating the direction of transcription. The figure was prepared using Protein Explorer with atomic coordinates deposited in the Protein Data Bank under accession number 1I50.
As shown in Figure 16.6, the DNA enters the assembly through a cleft formed by Rpb1 and Rpb2. The DNA is gripped by protein “jaws” but cannot pass straight through the molecule because of a protein “wall” and makes a right angle bend facilitating the addition of nucleotide triphosphates (NTPs). The NTPs enter through a “funnel” and pass through a pore to reach the active site for polymerization. Exit of the nascent messenger RNA chain is guided by several other structural elements, most notably by rudder, lid, and clamp elements. Most of these functional elements belong to the large Rpb1 and Rpb2 subunits. These subunits and the assignments of functional roles to portions of each are depicted in Figure 16.7.
16.8 Regulation by Chromatin-Modifying Enzymes
Chromatin-modifying enzymes regulate the accessibility of the DNA to transcription factors and the basal transcription machinery. Recall from Chapter 2 that histone tails undergo several different kinds of posttranslational modifications. The main targets of the modifications are amino groups situated at the end of lysine side chains (see Figure 2.13). Regulatory

396 16. Gene Regulation in Eukaryotes
FIGURE 16.7. Functional roles of the large subunits of RNA polymerase II: (a) Rpb1 subunit and (b) Rpb2 subunit. The figure was prepared using Protein Explorer with atomic coordinates deposited in the Protein Data Bank under accession number 1I50.
enzymes functioning as histone acetyltransferases (HATs) catalyze the addition of acetyl groups, while histone deacetylases (HDACs) do the opposite, catalyzing the removal of these groups. Acetyl groups are not the only ones added and removed from histone tails. Phosphoryl, methyl, and SUMO groups are added and removed as well, as was discussed in Chapter 2.
Transcriptionally inactive heterochromatin is tightly compacted. Sites where transcription initiation and regulation take place are inaccessible to the proteins responsible for these actions. In contrast, transcriptionally active euchromatin has a far more open shape, where transcription factors and the basal transcription machinery bind are accessible to the responsible proteins. The covalent addition of acetyl groups to the side chains reduces the net positive charge on the histone tails thereby weakening their attraction to the DNA strands. The addition of these groups counteracts the natural tendency for the chromatin fibers to fold into compact nucleosomal units making transcription difficult to impossible. Upon acetylation, the promoter sites become far more accessible to the transcription machinery.
The enzymes that mediate the modifications on the H3 and H4 tails are quite specific. This aspect, together with the presence of so many potential modification sites, has led to the suggestion that there is a histone code that determined whether a particular gene or cluster of genes is silenced or not.
An example of how this might work is supplied by data regarding the H3 lysine 9 (K9) site. As indicated in Figure 2.13 this is a site that can be either acetylated or methylated. The selection process is a competitive one. Acetylation at K9 blocks methylation at that site, and vice versa. The silencing
16.9 Multiprotein Complex Use of Energy of ATP Hydrolysis |
397 |
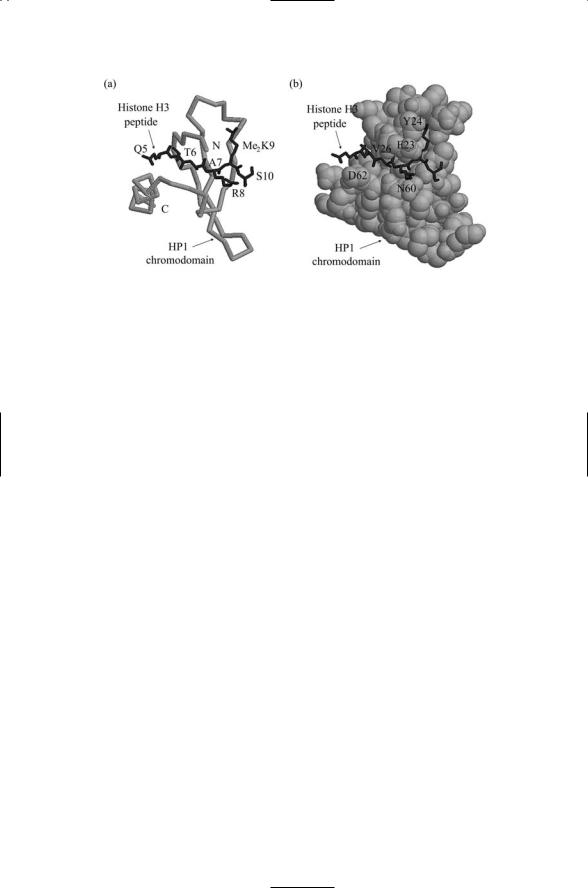
FIGURE 16.8. A peptide fragment of histone H3 doubly methylated on Lys9 (shown in black) complexed with the chromodomain of HP1 (shown in gray): Residues Gln5, Thr6, Ala7, and Arg8 of the H3 peptide form beta sheet interactions with residues Glu23, Tyr24, Val26, Asn60 and Asp62 of the chromodomain. The peptide residues are labeled in (a) while the chromodomain residues are labeled in (b). In
(a) the chromodomain backbone is highlighted, while in (b) a space-filled representation is presented. The atomic structures were determined using X-ray crystallography. The figure was prepared using Protein Explorer with atomic coordinates deposited in the Protein Data Bank under accession number 1KNA.
process for that site works in the following way. An HDAC acts on K9 to remove an acetyl group. Next, a histone methyltransferase-specific for that site catalyzes the covalent addition of a methyl group. Then, heterochromatin (adapte) protein 1 (HP1) binds to the methylated K9 site and blocks transcription. The binding of HP1 to the methylated histone tail is shown in Figure 16.8.
16.9Multiprotein Complex Use of Energy of ATP Hydrolysis
There are two basic kinds of chromatin modifiers. There are modifiers of chemical affinity between histones and DNA, and there are hydrolyzing enzymes that break and reform bonds between histones and DNA. The first group consists of enzymes such as the HATs and HDACs that act on the histone tails, covalently modifying them through the addition or subtraction of acetyl, methyl, or phosphoryl groups as just discussed. The second group encompasses several large multisubunit complexes that use the energy of ATP hydrolysis to disrupt interactions between histones and the DNA. There are three classes of ATP-dependent multisubunit complexes. Each contains at least one ATPase plus additional subunits. Some of the

398 16. Gene Regulation in Eukaryotes
TABLE 16.4. Three families of ATP-dependent SWI chromatin remodeling complexes: Abbreviations—ATP-dependent chromatin assembly and remodeling factor (ACF); chromatin accessibility complex (CHRAC); chromatin organization modifier (Chromo); Imitation SWI (ISWI); nucleosome remodeling factor (NURF); remodels the structure of chromatin (RSC); sucrose nonfermenting (SNF); switch (SWI); Yeast (y); Drosophila (d); human (h).
Family |
Complexes |
Recognition domain |
SWI/SNF |
ySWI/SNF, hSWI/SNF, yRSC |
Bromodomain |
ISWI |
dCHRAC, dNURF, dACF |
SANT domain |
Mi-2 |
Mi-2 |
Chromodomain |
|
|
|
complexes identified to date in various organisms are summarized in Table 16.4.
16.10Protein Complexes Act as Interfaces Between TFs and RNAP II
Chromatin modifying enzymes do not interact with the basal transcription machinery, but instead make it possible for that machinery to find and attach to the promoters. Other complexes then take over and further support transcription. These interfacing complexes link activators and enhancers to the basal machinery attached to the core promoter. These complexes function as interfaces integrating and conveying to RNA polymerase II and its suite of general transcription factors instructions embodied in the transcription factors.
The best studied of these interfacing complexes is the Mediator complex found in yeast. This complex consists of about 20 different proteins arranged into three modules. Figure 16.9 illustrates how this complex can serve as an interface between the core machinery and the transcription factors bound to activator and enhancer sites. As can be seen in this figure the mediator complex forms a bridge between the regulatory proteins and basal transcription machinery at the core promoter. This bridging action is especially important for proteins bound at distal enhancer sites brought into close proximity to the core promoter through bending and looping of the DNA.
The yeast mediator complex has a number of metazoan counterparts. These complexes each contain homologs of a number of the yeast subunits along with additional members specifically needed to deal with the metazoan regulatory pathways. Some of the metazoan complexes are quite large with more than a dozen members, while others are smaller composed of subsets of the larger complexes. As is the case for the Mediator, the physical dimensions are large enough to act as bridges. For example, the planar dimensions of the activation-recruited coactivator-large (ARC-L) complex

16.11 Alternative Splicing to Generate Multiple Proteins |
399 |
FIGURE 16.9. Yeast Mediator complex: Shown are the three Mediator modules— The Srb4 “head” domain, the Med9/10 “body” domain, and the Gal11/Sin4 “tail” domain, each named for prominent members of the module.
are 420 Å by 185 Å with distinct head, body, and tail regions formed by specific groups of subunits. The smaller coactivator required for Sp1 activation
(CRSP) complex, which consists of a subset of the ARC-L subunits, is still 360 Å by 145 Å. Another mammalian Mediator complex, called SMCC/TRAP (SRBand MED-containing cofactor complex/thyroid hormone receptor-associated protein), has about 25 members. These large mediator complexes attach to the RNAP II CTD and form holoenzymes with a total mass of about 1.5 MDa.
16.11Alternative Splicing to Generate Multiple Proteins
The remarkable efficiency of genetic coding is exemplified by the relatively modest increases in genome size in going from the yeast to the worm and the fly, and to vertebrates and man. A central feature in making this possible is the modular organization of the genome and the proteins so encoded. In higher eukaryotes, genes consist of short coding sequences, or exons, separated from one another by longer noncoding or intervening sequences, or introns. The exons are typically 50 to 400 nucleotides in length, while introns can be as long as 200,000 nucleotides. Whereas most yeast premRNAs do not contain introns, and those that do usually contain only a single one, in higher eukaryotes a single gene may contain as many as 50 introns. The introns are included in pre-mRNAs produced by transcription from the DNA templates. In splicing, introns situated in between pairs of flanking exons are removed. These intervening sequences are removed from pre-messenger RNAs (pre-mRNAs) to form mature mRNAs containing a selected set of exons that is used for protein synthesis.
400 16. Gene Regulation in Eukaryotes
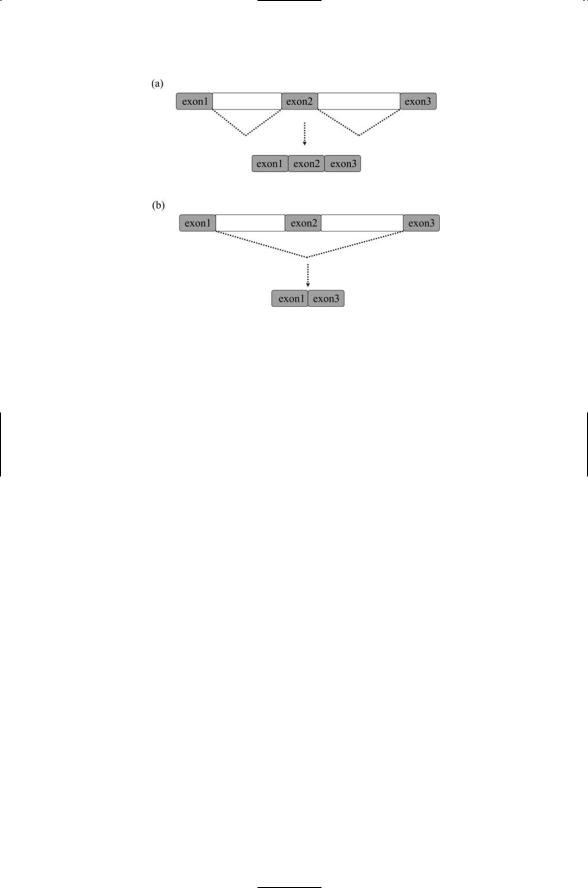
FIGURE 16.10. Alternative splicing of pre-messenger RNAs: Alternative splicing is an efficient method of generating multiple proteins from a single primary transcript in a controlled way. (a) The dotted lines indicate the intervening DNA sequences that are spliced out to produce a mature messenger RNA molecule containing three exons. (b) A different mix of exons is selected; the middle exon is skipped; that is, it is spliced out too, resulting in a mature mRNA molecule consisting of exon1 and exon3.
In alternative splicing, different sets of pre-mRNA sites are selected for splicing. This process can generate several different isoforms, or splice variants, of a single gene. By varying the mix of splice sites a variety of messenger RNAs (mRNAs) can be generated differing from one another by the presence of certain exons and the absence of others that have been skipped. This selection process is illustrated schematically in Figure 16.10. Alternative splicing generates a variety of tissue and compartment-specific proteins from a single gene; it is commonplace in the expression of genes that encode control layer proteins, especially those that function in the nervous and immune systems.
16.12Pre-Messenger RNA Molecules Contain Splice Sites
The pre-mRNA molecule contains exons, introns, and regulatory sequences that provide binding sites for the splicing machinery and regulatory proteins. Three sites are depicted in Figure 16.11. The first is the 5¢ site characterized by the presence of a binding sequence containing a guanine-uracil (GU) pair within a longer GURAGU-like sequence, where R is a purine.