

Molecular and Cellular Signaling - Martin Beckerman
.pdf14.15 p53 Halts Cell Cycle While DNA Repairs Are Made |
349 |
FIGURE 14.8. Growth signal stimulation of the transition from G1 phase to S phase: Activated Cdk4s and Cdk6s along with kinases activated by growth signals hyperphosphorylate the retinoblastoma protein (pRb). In response, the E2Fs dissociate from pRb and stimulate transcription of genes required for S-phase. Activation of cyclin E is associated with the passage to S phase from G1 phase. The transcription of cyclin E along with cyclin A and DNA polymerase is stimulated by the E2Fs. These actions are terminated when Cyclin A-Cdk2 build up and turn off the E2Fs transcriptional activities.
a key role in inactivating pRb. Phosphorylation of pRb by these cell cycle regulators and by the growth factor-activated kinases frees the E2Fs from their pRb inhibition and they can then carry out their cell cycle- progression-driving transcriptional activities (Figure 14.8).
Two noncontiguous domains, referred to as the A and B domains, collectively form a binding locus known as an A/B pocket that binds many proteins to pRb (Figure 14.9). For that reason, pRb and other members of its family are known as A/B pocket proteins. This importance of this interaction manifests itself by the occurrence of cancer-causing mutations in residues located in the A/B pocket. The cyclin cell cycle regulators are among the key proteins that bind to the A/B pocket. The cyclins, most notably cyclins D and E, recruit cyclin-dependent kinases to the A/B pocket and to a docking site located in the C terminus.
14.15p53 Halts Cell Cycle While DNA Repairs Are Made
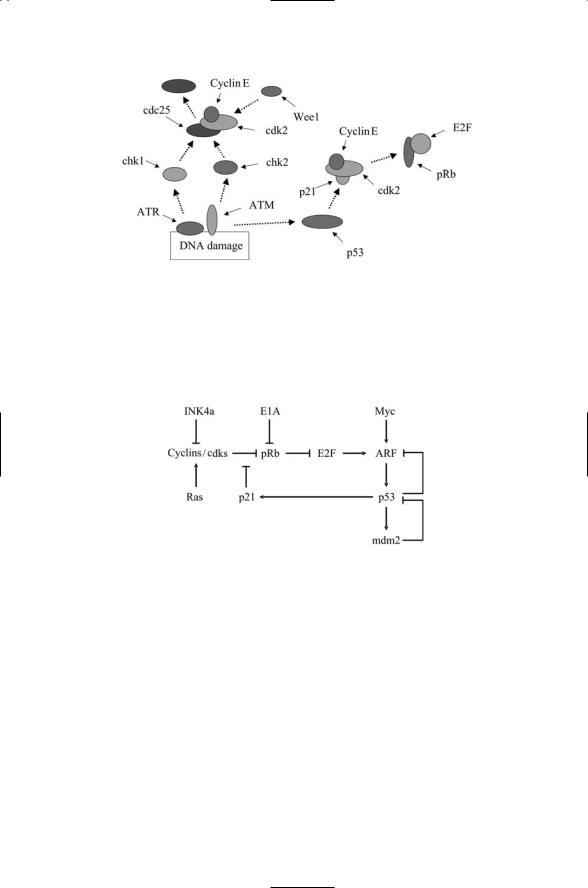
The integration of the G1/S DNA damage checkpoint pathway into the cell cycle occurs in the following way. Damage to DNA is sensed by the ATM/ATM and Rad3-related (ATR) kinases. When they detect damage to

350 14. Cancer
FIGURE 14.9. Binding of an E2F peptide fragment to the pRb pocket: Shown in the figure is a 18-residue fragment derived from the transactivation domain of E2F (residues 410–427) in a complex with the A/B pocket domain of pRb. The figure was generated using Protein Explorer with atomic coordinates deposited in the Brookhaven PDB under accession number 1N4M.
DNA they phosphorylate a pair of kinases called checkpoint 1 (Chk1) and checkpoint 2 (Chk2). The Chk1 and Chk2 kinases phosphorylate a protein phosphatase known as cell division cycle 25 (Cdc25). The Cdc25 protein phosphatase is a cyclin-cdk activator. It is active when it is in a dephosphorylated state and is inactivated and degraded when it is phosphorylated. Phosphorylation of Cdc25 by the checkpoint kinases inactivates the phosphatase and tags it for proteolysis, leading to phosphorylation and inactivation (arrest) of the cyclin-cdks through actions of another protein kinase called Wee1.
When damage to the cellular DNA is sensed, ATM, ATR, and also DNAPKcs convey that information to p53 by phosphorylating the molecule at sites specific to the type of damage sensed. In response to these signals, p53 acts as a transcription factor and upregulates a number of proteins. One of these, p21, halts the progression through the cell cycle until DNA repairs can be made. The p21 protein upregulated by p53 is a universal regulatory subunit of cyclin-cdks. When p21 is activated it binds to the Cyclin E/Cdk2 complex, resulting in Cdk2 inactivation. The Cdk2s cannot phosphorylate pRb, which then becomes hypophosphorylated and inhibits the E2Fs (Figure 14.10) thereby halting progression from G1 to S phase. If the damage is regarded as unrepairable, p53 functions in a second different way. It triggers an apoptosis circuit that targets the cell for suicide.
14.16p53 and pRb Controllers Central to Metazoan Cancer Prevention Program
The p53, pRb, and connecting signaling proteins and other key regulators form a circuit that regulates the progression through the cell cycle. Proper operation of this circuit is central to a cell’s cancer prevention program. As
14.16 p53 and pRb Controllers |
351 |
FIGURE 14.10. Cell cycle arrest triggered by DNA damage checkpoint signals: ATR and ATM phosphorylate Chk1 and Chk2, which then phosphorylate the protein phosphatase Cdc25 thereby inactivating it. Cdk2 is phosphorylated by the protein kinase Wee1 and its actions are arrested. ATM signals through p53 to activate p21, which arrests entry into S phase by inhibiting the transcriptional activities of the E2Fs.
FIGURE 14.11. Controller circuit coordinating p53 and pRB activities: Mitogenic signals are relayed into the cell via Ras, Ink4a and c-Myc. Pointed arrows denote stimulatory influences and flat-headed arrows denote repressive influences.
shown in Table 14.1, mutations in p53 or pRb leading to malfunctions in the circuitry are found in most human cancers. A simplified representation of this circuitry highlighting some of the most crucial protein-protein interactions is depicted in Figure 14.11. INK4a, Ras and c-Myc relay mitogenic (growth and proliferation) signals. These signals converge upon ARF which sits just upstream of p53. As shown in the figure, when it is activated, ARF stimulates p53 activity. It does so by disrupting mdm2’s inhibition of p53. Because of its key location, ARF mutations are encountered in a host of human cancers. Another prominent gene encountered in many cancers is the adenovirus early region 1A (E1A) gene. This gene product interacts with a variety of control proteins including pRb. When E1A binds pRb, it

352 14. Cancer
disrupts formation of pRb-E2F heterodimers and thus promotes establishment of cancer cell cycles resembling those produced by mutations in pRb itself.
The pRb and p53 work together. Early in the cell cycle, pRb inhibits the E2Fs, and Mdm2 keeps p53 at a low level of activity. When pRb becomes hyperphosphorylated its block on the E2F is relieved, ARF is stimulated and it, in turn, stimulates p53 so that as the cell enters S phase, p53’s surveillance activities are stepped up. If p53 detects DNA damage it signals p21, which blocks the cyclin/cdk complexes from hyperphosphorylating pRb. The retinoblastoma protein then inhibits the E2Fs while the repairs to DNA are carried out.
The ATM/ATR and DNA-PKcs proteins activate p53 by stimulating proteolysis of the Mdm2 inhibitor. Under normal cellular conditions, the Mdm2 protein represses the checkpointing and apoptosis promoting activities of p53. The Mdm2 protein acts as a p53-specific, E3 ubiquitin ligase. It suppresses the transcriptional actions of p53 by tagging it for proteolytic destruction and keeping its expression levels low. One of the genes turned on by p53 is the Mdm2 gene. This activity leads to the formation of an autoregulatory loop in which p53 regulates Mdm2 at the level of transcription, and Mdm2 regulates p53 at the level of its activity. The Mdm2p53 system is balanced through these mutual dependencies so that the appropriate signals can activate p53. These signals dislodge Mdm2 from p53, leading to the transcriptional activation of p53.
14.17p53 Structure Supports Its Role as a Central Controller
As might be expected from the appellation “controller,” the p53 and pRb proteins receive input signals from many proteins and in turn influence many other proteins. The first of these, p53, functions as a transcription factor while the second protein, pRb, acts through the E2F family of transcription factors. In the language of networks, p53 and pRb operate as highly connected nodes, and therefore it is not surprising that disabling these proteins through mutations has such strong negative consequences. The signals convergent upon p53 take the form of phosphorylations and acetylations of specific residues. Some of the best characterized of the sites of phosphorylation and acetylation are depicted in Figure 14.12.
The flexibility in the chain connecting the core unit to the DNA-binding domain allows the p53 molecule to contact the DNA in any of a number of ways. This property is illustrated in Figure 14.13 where three DNA-binding domains bind the DNA molecule, each in a slightly different way. The most frequent mutations in p53 found in cancer cells are all situated in the DNAbinding domain. Of these, five involved arginine residues and one a glycine residue. The leading mutation is to Arg248, which mediates the direct

14.17 p53 Structure Supports Its Role as a Central Controller |
353 |
FIGURE 14.12. Organization of the p53 protein: Shown in the upper part of the figure is the domain structure of the p53 monomer. It contains an N-terminal transactivation (TA) domain, a proline-rich (PR) region, a DNA-binding domain, and in its C- terminal region a nuclear export sequence (NES), tetramerization (Tetra) domain and a negative regulatory (NR) domain. Expanded depictions of some of the key regulatory regions are shown in the lower part of the figure. The specific amino acid residues that are phosphorylated (P), acetylated (A) or sumoylated (S) are shown. Abbreviations: serine (S), threonine (T), lysine (K).
FIGURE 14.13. p53-DNA binding: The DNA-binding domains of the p53 protein are depicted as ribbons while the dsDNA molecule is shown in a space-filled model. The figure was generated using Protein Explorer with atomic coordinates deposited in the Brookhaven PDB under accession numbers 1TUP and 1TSR.

354 14. Cancer
binding of the p53 molecule to the minor groove of DNA. Another often mutated residue, Arg273, makes contact with the backbone phosphate. The other residues help stabilize the interface between the p53 and DNA surfaces.
14.18 Telomerase Production in Cancer Cells
The ends of chromosomes are capped by telomeres, a series of TTAGGG repeats and associated proteins, and terminated at the very end by a lassolike structure called a T-loop. Telomeres are shortened by about 100 base pairs every cell division. When the capping structure is degraded sufficiently the cell’s DNA repair machinery is able to sense the ends of the DNA molecules and interprets the ends as double strand breaks. If the cell keeps dividing several negative outcomes become possible. These include degradation, chromosome recombination leading to loss of genetic information, aberrant rearrangements of chromosomes, and genomic instability. To avoid such dangerous situations the cell ceases to divide after several kilo base pairs of telomere are lost and instead enters a nondividing stage called senescence.
Several proteins associate with the telomeres. One of these is the (human) telomerase reverse transcriptase (hTERT), which catalyzes the addition of multiple TTAGGG repeats at the ends of chromosomes and protects them from the DSB repair machinery. Among the others are two proteins, named telomeric repeat binding factors 1 and 2 (TRF1 and TRF2), that bind the TTAGGG repeats. In addition, a number of double strand break repair proteins form complexes with the TRFs and the telomerase components (the hTERT catalytic subunit and a human telomerase RNA (hTR) that contains the template for adding telomeric repeats). This grouping includes members of the Rad50/Mre11/NBS1 double strand break repair complex, and Ku86 and DNA-PKcs involved in NHEJ. Members of these complexes are thought to convey prosenescence signals to the p53 circuitry when telomeres become shortened.
Signals sent through p53 and pRb ensure that the cell ceases to divide. In response to critical telomere shortening, production of regulators of p53 and pRb activity such as ARF, p21, and Ink4a is increased. The pRb protein becomes hypophosphorylated and binds E2F thereby suppressing proliferation. In more detail, ARF suppresses Mdm2 leading to the release of p53 from Mdm2 inhibition; p53 stimulates p21, which contributes to hypophosphorylation of pRb. The Ink4a protein contributes to pRb hypophosphorylation by suppressing the Ckd4/6 inhibition of pRb.
Cancer cells avoid senescence by increasing the production of the telomerase chromosome-capping enzyme. This activity greatly expands the number of cell divisions possible, effectively immortalizing the cells. The cells avoid passing into senescence, and instead continue to divide and

References and Further Reading |
355 |
progress towards more lethal states. Mutations in the p53 and pRb that disable these proteins can contribute to the immortalization by disrupting the conveyance of prosenescence signals.
References and Further Reading
MMPs
McCawley LJ, and Matrisian LM [2001]. Matrix metalloproteinases: They’re not just for matrix anymore! Curr. Opin. Cell Biol., 13: 534–540.
Nagase H, and Woessner JF, Jr [1999]. Matrix metalloproteinases. J. Biol. Chem., 274: 21491–21494.
Stamenkovic I [2003]. Extracellular matrix remodeling: The role of matrix metalloproteinases. J. Pathol., 200: 448–464.
Growth Factor Signaling
Birchmeier C, et al. [2003]. MET, metastasis, motility and more. Nature Rev. Mol. Cell Biol., 4: 915–925.
Blume-Jensen P, and Hunter T [2001]. Oncogenic kinase signaling. Nature, 411: 355–365.
Downward J [2003]. Targeting Ras signaling pathways in cancer therapy. Nature Rev. Cancer, 3: 11–22.
Growth Factor Receptor, and Adhesion Molecule Cooperativity
Comoglio PM, Boccaccio C, and Trusolino L [2003]. Interactions between growth factor receptors and adhesion molecules: Breaking the rules. Curr. Opin. Cell Biol., 15: 565–571.
Conacci-Sorrell M, Zhurinsky J, and Ben-Ze’ev A [2002]. The cadherin-catenin adhesion system in signaling and cancer. J. Clin. Invest., 109: 987–991.
Hood JD, and Cheresh DA [2002]. Role of integrins in cell invasion and migration.
Nature Rev. Cancer, 2: 91–100.
Developmental Pathways
Fearnhead NS, Britton MP, and Bodmer WF [2001]. The ABC of APC. Human Mol. Genet., 10: 721–733.
Giles RH, van Es JH, and Clevers H [2003]. Caught up in a Wnt storm: Wnt signaling in cancer. Biochim. Biophys. Acta, 1653: 1–24.
Massagué J, Blain SW, and Lo RS [2000]. TGFb signaling in growth control, cancer and heritable disorders. Cell, 103: 295–309.
Peifer M, and Polakis P [2000]. Wnt signaling in oncogenesis and embryogenesis— A look outside the nucleus. Science, 287: 1606–1609.
Taipale J, and Beachy PA [2001]. The Hedgehog and Wnt signalling pathways in cancer. Nature, 411: 349–354.
DNA Repair Mechanisms
Critchlow SE, and Jackson SP [1998]. DNA end-joining: From yeast to man. Trends Biochem. Sci., 23: 394–398.

356 14. Cancer
De Laat WL, Jaspers NGL, and Hoeijmakers JHJ [1999]. Molecular mechanisms of nucleotide excision repair. Genes Dev., 13: 768–785.
Hoeijmakers JHJ [2001]. Genome maintenance mechanisms for preventing cancer. Nature, 411: 366–374.
Kanaar R, Hoeijmakers JHJ, and van Gent DC [1998]. Molecular mechanisms of DNA double-strand break repair. Trends Cell Biol., 8: 483–489.
Lindahl T, and Wood RD [1999]. Quality control by DNA repair. Science, 286: 1897–1904.
Yang W [2000]. Structure and function of mismatch repair proteins. Mutation Res.— DNA Repair, 460: 245–256.
Double-Strand-Break Repair
D’Amours D, and Jackson SP [2002]. The Mre11 complex:At the crossroads of DNA repair and checkpoint signaling. Nature Rev. Mol. Cell Biol., 3: 317–327.
D’Andrea AD, and Grompe M [2003]. The Fanconi anaemia/BRCA pathway.
Nature Rev. Cancer, 3: 23–34.
Hopfner KP, et al. [2002]. The Rad50 zinc-hook is a structure joining Mre11 complexes in DNA recombination and repair. Nature, 418: 562–566.
Leuther KK, et al. [1999]. Structure of DNA-dependent protein kinase: Implications for its regulation by DNA. EMBO J., 18: 1114–1123.
Song BW, and Sung P [2000]. Functional interactions among yeast Rad51 recombinase, Rad52 mediator, and replication protein A in DNA strand exchange. J. Biol. Chem., 275: 15895–15904.
Yang HJ, et al. [2002]. BRCA2 function in DNA binding and recombination from a BRCA2-DSS1-ssDNA structure. Science, 297: 1837–1848.
The ATM Protein
Bakkenist CJ, and Kasten MB [2003]. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation, Nature, 421: 499–506.
Shiloh Y [2003]. ATM and related protein kinases: Safeguarding genome integrity.
Nature Rev. Cancer, 3: 155–168.
The Cell Cycle, E2Fs, and the Retinoblastoma Protein
Dyson N [1998]. The regulation of E2F by pRb-family proteins. Genes Dev., 12: 2245–2262.
Harbour JW, et al. [1999]. cdk phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1. Cell, 98: 859–869.
Malumbres M, and Barbacid M [2001]. To cycle or not to cycle: A critical decision in cancer. Nature Rev. Cancer, 1: 222–231.
Muller H, and Helin K [2000]. The E2F transcription factors: Key regulators of cell proliferation. Biochim. Biophys. Acta., 1470: M1–M12.
Muller H, et al. [2001]. E2Fs regulate the expression of genes involved in differentiation, development, proliferation, and apoptosis. Genes Dev., 15: 267–285.
Sherr CJ [2000]. The Pezcoller Lecture: Cancer cell cycles revisited. Cancer Res., 60: 3689–3695.
Weinberg RA [1995]. The retinoblastoma protein and cell-cycle control. Cell, 81: 323–330.

Problems 357
The p53 Protein
Espinosa JM, and Emerson BM [2001]. Transcriptional regulation by p53 through intrinsic DNA/chromatin binding and site-directed cofactor recruitment. Mol. Cell, 8: 57–69.
Sherr CJ [2001]. The INK4a/ARF network in tumor suppression. Nature Rev. Mol. Cell Biol., 2: 731–737.
Vogelstein B, Lane D, and Levine AJ [2000]. Surfing the p53 network. Nature, 408: 307–310.
Vousden KH, and Lu X [2002]. Live or let die: The cell’s response to p53. Nature Rev. Cancer, 2: 594–604.
Telomere Maintenance
Blackburn EH [2001]. Switching and signaling at the telomere. Cell, 106: 661–673. Chan SWL, and Blackburn EH [2002]. New ways to make ends meet: Telomerase,
DNA damage proteins and heterochromatin. Oncogene, 21: 553–563.
De Lange T [2002]. Protection of mammalian telomeres. Oncogene, 21: 532–540. Itahana K, Dimri G, and Campisi J [2001]. Regulation of cellular senescence by p53.
Eur. J. Biochem., 268: 2784–2791.
Maser RS, and DePinho RA [2002]. Connecting chromosomes, crisis and cancer. Science, 297: 565–569.
Problems
14.1Make a list of the ways a cancer cell differs from a normal one. Identify the signaling proteins that promote the altered response and how these proteins are altered to promote cancer for each entry in the list. For example, mutations in the Ras proteins that leave the GTPase stuck in the on position contribute to aberrant growth signaling that cannot be turned off.
14.2Cancer is a multistep process. Arrange the entries in the list generated in Problem 14.1 in a temporal order with early acting changes at the top and late acting changes at the bottom. Which kinds of alterations promote metastasis? Which ones help generate genome instabilities and chromosome abnormalities?

15
Apoptosis
Kerr, Wyllie, and Currie coined the term apoptosis in an article that appeared in the British Journal of Cancer in 1972. The term was taken from the Greek and has the meaning of “falling off” as in the dropping of petals from flowers, and leaves from trees, which occur in a programmed manner every autumn. Apoptosis, or programmed cell death, is widespread during animal development where it is used to sculpt and refine tissues and organs. Apoptosis makes possible the regression of tadpole tails, permitting the emergence of an adult tailless form. It is used to remove larval organs in insects, and to sculpt digits—fingers and toes—from undelineated limb buds in mammals.
Apoptosis is widely encountered during development of the nervous system. Some neurons are only transiently formed, and once their task is completed they are removed. In many parts of the nervous system, cells are initially overproduced. This overproduction ensures that there is adequate input to target neurons during the initial phase of the nervous system connection. Excess cells are removed by numerically matching preand postsynaptic structures. The initial set of neural connections is refined, and cells that are inappropriately wired are removed. In some neural populations, most of the cells are removed in order to produce a set of precise neural connections.
Apoptosis is an essential ingredient in the operation of the immune system. Cells of the immune system such as B cells die off as they age and when they are no longer needed. Virally infected cells, and damaged cells that cannot be repaired, are targeted for apoptosis to prevent their harming undamaged cells in the tissue or organ where they reside. The same strategy is used by the immune system to rid the body of cancer cells.
Apoptosis is different from necrosis. The plasma membrane does not rupture in apoptosis as it does in necrosis, but instead cellular components are degraded and packaged, and then digested. During apoptosis cells undergo an orderly sequence of morphological changes. These changes include cell shrinkage, chromatin condensation, DNA fragmentation, and membrane blebbing, in which membrane-wrapped pieces of cell boil off of
359