

8.2. Механизм ориентации - влияние заместителей на выбор
места электрофильной атаки
Электронодонорные группы активируют все положения кольца, т.е. орто-, пара- и мета-положения становятся более реакционноспособными, чем любое положение в бензоле. Однако они активируют орто- и пара-положения сильнее, чем мета-положение.
Электроноакцепторные группы (кроме галогенов) дезактивируют все положения кольца, но особенно сильно - орто- и пара-положения.
Галогены составляют особую группу ориентантов, которая будет рассмотрена отдельно.

Следовательно, любая группа: - и активирующая, и дезактивирующая - самое сильное влияние оказывает на орто- и пара-положения. Это обусловлено тем, что именно в орто- и пара-положениях по отношению к углероду, присоединившему электрофил, возникает положительный заряд.
Поскольку медленной стадией реакции электрофильного замещения является стадия образования о-, м-, п--комплексов (карбокатионов), соотношение, в котором они образуются в этой медленной стадии, определяет количества о-, м-, п-изомерных продуктов реакции. Известно, что чем больше распределён заряд в карбокатионе, тем он устойчивее и тем быстрее образуется. Следовательно, главное направление реакции электрофильного замещения осуществляется через образование наиболее устойчивых карбокатионов (-комплексов).
Рассмотрим влияние электронодонорных групп на устойчивость образующихся в медленной стадии карбокатионов.
1). Влияние группы СН3–, проявляющей электронодонорный индукционный эффект.

Группа -СН3 подает электроны ко всем атомам кольца (+I - эффект), но наибольшее влияние она оказывает на тот атом, с которым непосредственно связана. Среди граничных структур I-III, изображающих орто--комплекс, есть структура I, в которой метильная группа подает электроны на углеродный атом, несущий положительный заряд, и за счёт этого в ней в большей степени, чем в II и III, распределяется положительный заряд. Структура I более устойчива, вклад её в резонансный гибрид наибольший. Аналогичен вклад структуры IV в пара--комплекс.
В мета--комплексе группа -СН3 не проявляет в такой же степени способность стабилизировать его: ни в одной из граничных структур VII-IX она не связана с положительно заряженным углеродным атомом.
Следовательно, из трех активированных комплексов наиболее устойчивыми являются о-, п--комплексы, в которых положительный заряд находится на атоме, несущем группу -CH3. Это значит, что энергии активации образования о-, п--комплексов ниже, чем энергия активации образования м--комплекса, продукты о-, п-замещения образуются в большем количестве, чем м-продукт.

2). Несколько другой тип стабилизации -комплекса характерен для групп, содержащих гетероатом атом с неподеленной электронной парой: -ОН, -ОR, -NН2, имеющих неподеленные электроны (р--сопряжение) и -CH=CH2; -С6Н5, содержащих -электроны (--сопряжение). Группы -ОН, -ОR, -NН2, -NНR, -NR2 проявляют электроноакцепторный индукционный эффект. Это означает, что атомы азота и кислорода, соединённые с углеродом ароматического кольца, оттягивают на себя электроны -связи вследствие большей электроотрицательности азота и кислорода по сравнению с углеродом. Однако эффект р--сопряжения этих групп преобладает над электроноакцепторным индукционным эффектом.
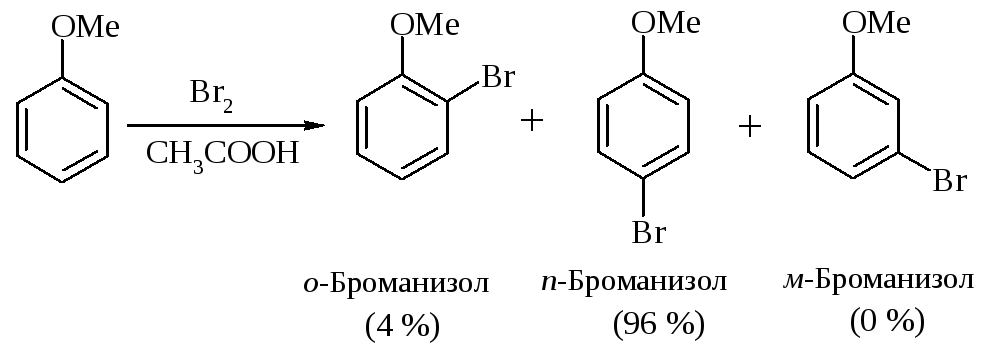
Рассмотрим строение -комплексов, образующихся при электрофильном замещении в анизоле.

-Комплекс, образующийся при атаке в орто-положение, представляет собой резонансный гибрид структур I-III, в которых положительный заряд распределен между атомами углерода, а также структуры IV, в которой положительный заряд находится на атоме кислорода. Структура IV особенно устойчива, поскольку каждый атом имеет завершённую электронную оболочку. Подобная картина возникает и при атаке в пара-положение.
Но в случае мета-атаки структура, подобная IV или VIII, в изображении -комплекса отсутствует. Следовательно, орто- и пара--комплексы более устойчивы, а значит, замещение идёт преимущественно в пара- и орто-положения.
Теперь сравним устойчивость -комплексов, образующихся при атаке в орто-, мета- и пара-положения бензольного кольца, содержащего электроноакцепторную группу.

-Комплексы, образующиеся при атаке в орто- пара- и мета-положения, менее устойчивы и образуются медленнее, чем карбокатион, возникающий при взаимодействии электрофила с бензолом. Это означает, что электроноакцепторные группы дезактивируют бензол в реакциях электрофильного замещения.
Сравнение граничных структур I-III, IV-VI и VII-IX приводит нас к выводу: из трёх -комплексов наименее устойчивы орто- и пара--комплексы, так как в структурах I и IV положительные заряды расположены на соседних атомах, вклад этих структур можно не учитывать, заряд в орто- и пара--комплексах распределён в меньшей степени, чем в мета--комплексе. Следовательно, замещение в орто- и пара-положения идёт медленнее, чем в мета-, и основным продуктом является мета-замещённое соединение.

Рассмотрение механизма ориентации показывает, что все электроноакцепторные (кроме галогенов) группы являются дезактивирующими мета-ориентантами, а все электронодонорные группы - активирующими орто-, пара-ориентантами (табл. 8.1).
Почему галогены ведут себя необычным образом? Благодаря резонансному эффекту (эффекту сопряжения) галогены являются орто-, пара-ориентантами.

Резонанс такого типа требует перекрывания 2р-орбиталей атома углерода с 3р-орбиталями атома хлора. Так как в сопряжение вовлекаются электроны различных квантовых уровней, это сопряжение не будет так эффективно, как сопряжение электронов углерода и азота, углерода и кислорода.
Таблица 8.1