

2.6. Методы разделения энантиомеров
Наиболее универсальным методом разделения смеси энантиомеров является химический. Он состоит в следующем. Рацемическую смесь энатиомеров обрабатывают оптически активным соединением, в результате образуется смесь диастереомеров, обладающих разными физическими константами.
Например, при реакции рацемической кислоты (R,S)–А с оптически активным основанием (R)–B образуется смесь солей:
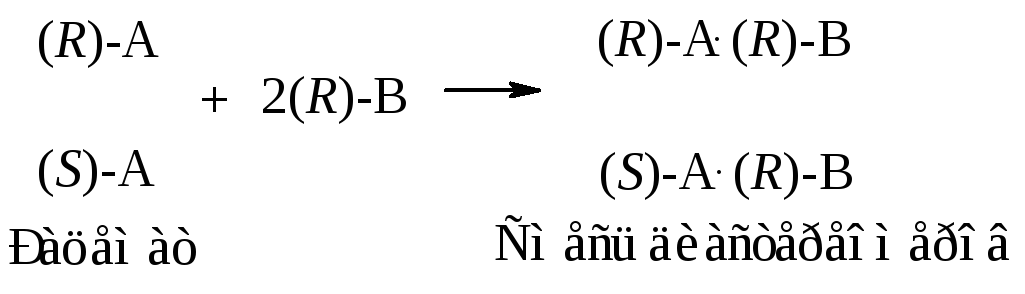
Из полученных таким образом индивидуальных диастереоизомеров выделяют энантиомеры.
3. Циклоалканы
Углеводороды, которые содержат кольца, состоящие из углеродных атомов, связанных между собой простыми связями, называются алициклическими или циклоалканами (циклопарафинами). Ниже приведена полная и сокращенная запись структурных формул первых четырех представителей ряда циклоалканов.
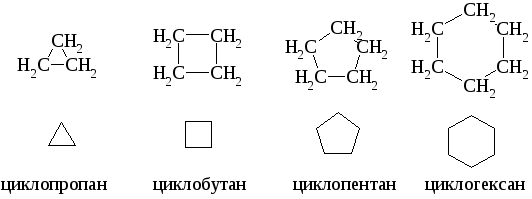
3.1 Номенклатура. Изомерия
Названия циклоалканов образуются добавлением приставки цикло- к названию линейного алкана с тем же числом атомов углерода. В алициклических соединениях известны следующие виды изомерии: структурная (изомерия, связанная с различной величиной цикла, различным строением и положением в цикле боковых цепей), пространственная (геометрическая или цис-, транс-изомерия, обусловленная различным расположением групп относительно плоскости кольца) и оптическая (энантиомерия).
Примеры структурных изомеров C6H12:
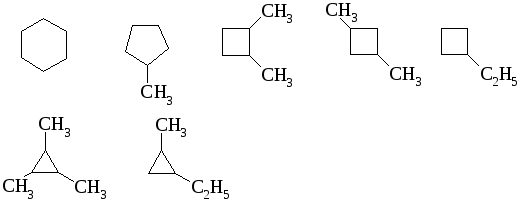
Примеры геометрических изомеров:
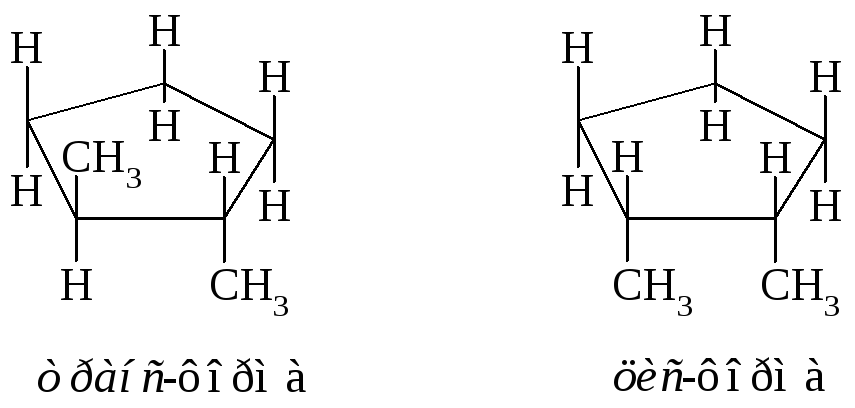
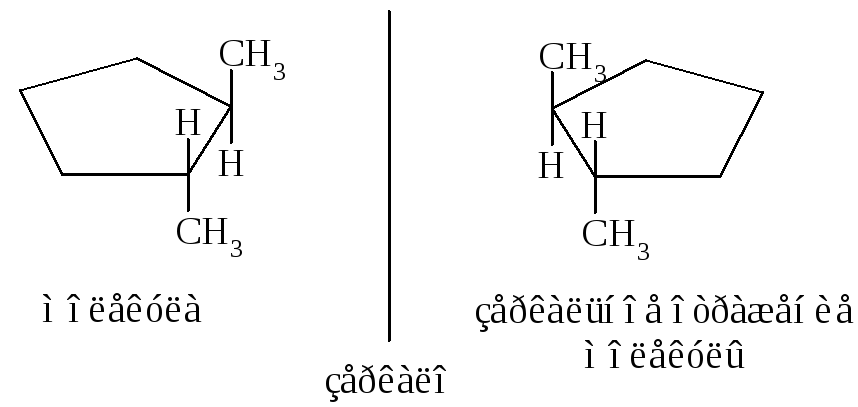
Примеры энантиомеров:
3.2. Физические свойства
Физические свойства циклоалканов сходны со свойствами соответствующих ациклических углеводородов, хотя температуры кипения и плавления циклических соединений немного выше. Циклоалканы неполярные или малополярные соединения, поэтому они хорошо растворимы в неполярных растворителях, таких как четыреххлористый углерод, эфир, и нерастворимы в сильно полярном растворителе - воде.
3.3. Строение
Теория напряжения А. Байера. В 1885 г. профессор Мюнхенского университета А.Байер предложил теорию, объясняющую некоторые аспекты химии циклических соединений. Часть его теории, рассматривающая способность к раскрытию малых циклов, общепринята и сегодня, хотя сейчас она излагается с других, современных позиций.
Байер рассуждал следующим образом. Когда атом углерода связан с четырьмя другими атомами (sp3-гибридизация), между каждыми двумя связями образуется угол 109о28.

Предполагалось, что в молекулах циклоалканов атомы углерода являются вершинами правильных плоских многоугольников. Циклопропан представляет собой плоский правильный треугольник с углом между связями С–С, равным 60о.
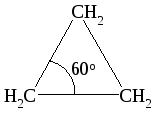
В циклопропане две связи у каждого из атомов углерода не могут образовать нормальный тетраэдрический угол 109о28, угол между ними сжат до 60о. Такое отклонение от нормального тетраэдрического угла делает эту молекулу “напряженной” и, следовательно, неустойчивой по сравнению с молекулами с тетраэдрическими углами.
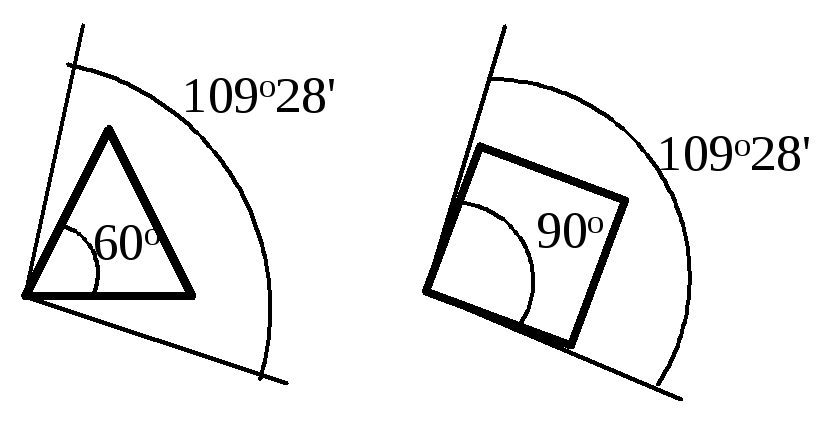
Циклоропан вступает в реакции с раскрытием кольца, поскольку при этом снимается угловое напряжение и образуются более устойчивые ациклические соединения. Чем больше отклонение от нормального угла 109о28, тем более “напряженной” является молекула: для циклопропана отклонение составляет 1/2(109о28 - 60о)= 24о44, а для плоского циклобутана -1/2(109о28 - 90о)= 9о44.
Поскольку искажение углов наиболее значительно в циклопропане, то он является более “напряженным”, более неустойчивым, более склонным к реакциям, протекающим с раскрытием кольца. Углы в правильном плоском пятиугольнике весьма близки к тетраэдрическим (108о), и поэтому циклопентан практически свободен от углового напряжения.
Углы в правильном плоском шестиугольнике (120о) несколько превышают тетраэдрические, на основании чего Байер предположил (ошибочно), что в циклогексане должно быть некоторое напряжение, а при переходе к циклогептану, циклооктану и т. д. отклонения от угла 109о28 будут увеличиваться, вследствие этого молекулы будут становиться все более напряженными.
Как согласуется теория Байера с фактами?
Полезную информацию об относительной устойчивости органических соединений дают теплоты сгорания веществ. Теплота сгорания - это количество теплоты, которое выделяется при сгорании одного моля вещества до углекислого газа и воды. Рассчитано, что для алифатических соединений сгорание метиленового звена -СН2- дает 659 кДжмоль -1 .
-СН2- О2 СО2 Н2О тепло
В основе классификации циклов, первоначально кажущейся произвольной, лежит зависимость между размером кольца и его устойчивостью.
Если циклопропан и циклобутан выделяют при сгорании больше энергии в расчете на СН2-группу, чем ациклические соединения (соответственно на 38,5 и 27,3 кДж/моль), то это означает, что они содержат больше энергии (см. табл. 3.1). Тогда в соответствии с теорией напряжения Байера циклопропан и циклобутан менее устойчивы по сравнению с ациклическими соединениями, и это обусловливает их склонность к реакциям с раскрытием кольца. В соответствии с теорией Байера циклы большие, чем циклогексан и циклобутан, должны быть неустойчивыми и иметь высокие теплоты сгорания. Но из таблицы 3.1 видно, что теплоты сгорания нормальных и средних циклов в расчете на одну СН2-группу мало отличаются от теплоты сгорания СН2-группы ациклических углеводородов, а для больших циклов - теплота сгорания СН2-группы практически равна этой величине. В противоположность теории Байера ни одна из этих систем не обладает меньшей устойчивостью по сравнению с ациклическими соединениями и не обнаруживает тенденции вступать в реакции раскрытия цикла подобно циклопропану.
Таблица 3.1