

Ovru-all
.pdfчислу станів з даною енергією. Ламана лінія символізує випадкове блукання в просторі конфігурацій при послідовних змінах однієї з координат, що характеризують мікростан системи, згідно з вищеназваними алгоритмами. Ця ламана найчастіше проходить через найвигідніші з ентропійно-енергетичної точки зору макростани (не дуже велика енергія і велика кількість мікростанів з такою енергією – мінімальній енергії відповідає тільки один мікростан й відповідно нульова ентропія). При цьому більшість конфігурацій макроскопічної системи взагалі не реалізується. Тому середні значення фізичних величин можна обчислити з використанням лише тих конфігурацій, які були реалізовані в результаті випадкового блукання в процесі моделювання. Чим довше відбувається це блукання, тобто чим більше воно захопило конфігурацій (або, як зазвичай кажуть, чим краще набрана статистика), тим точніше будуть обчислення.
1.2.3. Алгоритм Метрополіса й алгоритм термостата
Метод Метрополіса був сформульований в його роботі з розрахунку рівняння стану рідини, що складається з твердих дисків [8]. Сутність методу полягає в генерації набору послідовних конфігурацій системи. На кожному кроці цього процесу за наявною попередньою конфігурацією будується наступна, що відрізняється від попередньої зміною одного зі ступенів вільності. Нова конфігурація може бути прийнята або не прийнята в ансамбль у відповідності до процедури статистичного випробування. Якщо за результатом статистичного випробування нова конфігурація відкинута, то в ансамбль на даному кроці ще раз включається попередня конфігурація. Таким чином, в залежності від важливості, вага даної конфігурації в ансамблі може зростати.
Визначивши спосіб блукання фазовим простором системи, необхідно задати імовірності переходу між двома конфігураціями на даному кроці блукання. При цьому треба виходити з того міркування, що в граничному випадку великого числа блукань система переходить у рівноважний стан. Це означає, що середнє від будь-якої фізичної величини A за Марковим часом обчислюється за формулою
N
<A>N=(1/N) ∑ A(ti ) . (1.13)
i=1
де ti – i-й момент (крок) часу, в який зроблено вимірювання величини A, в граничному випадку великого числа N вимірювань має пере-
21
ходити в статистичне середнє від цієї величини за канонічним ансамблем.
Процедура статистичного випробування в методі Метрополіса полягає в тому, що нова конфігурація приймається в ансамбль з імовірністю, що дорівнює відношенню Гіббсових вагів (експонент Больцмана) нової та старої конфігурації. Наприклад, для моделі Ізінга, стара і нова конфігурації відрізняються перекиданням одного спіна. Імовірність прийняття нової конфігурації
р(1→2)=[ехр(–E2/kT)]/[ехр(–E1/kT)]=ехр[(E1–E2)/(kT)] (1.14)
у випадку E2>E1, і р(1→2)=1, якщо E2<E1 (E2 і E1 – енергії, відповідно, нової і старої конфігурації). Оскільки імовірність наступної конфігурації визначається лише попередньою конфігурацією і не залежить від передісторії, послідовні конфігурації утворюють ланцюг Маркова. Число реалізованих кроків такого процесу ми будемо називати часом Маркова.
Суттєвою і вельми відповідальною складовою алгоритмів Метрополіса і термостата є генерація псевдовипадкових чисел. Ці числа звуться псевдовипадковими, а не випадковими, оскільки будь-який запрограмований на ЕОМ генератор таких чисел має кінцевий період, після якого ці числа починають повторюватись. Це відбувається через скінченність числа десятинних розрядів в представленні дійсних чисел на ЕОМ. Якість генератора випадкових чисел істотно впливає на точність обчислень за методом Монте-Карло.
Головна властивість алгоритму Метрополіса полягає в тому, що він «заганяє» систему в область найбільш імовірних значень у фазовому просторі. Більша частина конфігурацій ігнорується при побудові Маркового процесу, і усереднення фізичних величин здійснюється лише за найбільш імовірними конфігураціями.
В якості вихідної конфігурації при побудові Маркового ланцюга можуть використовуватись будь-які значення змінних, що відповідають ступеням вільності системи. В принципі, від їх вибору результат не має залежати: система в стані рівноваги «забуває» про історію встановлення рівноваги. Однак, при реальних розрахунках від вдалого вибору вихідної конфігурації може залежати тривалість досягнення типових рівноважних конфігурацій, за якими можна здійснювати вимірювання. Виняток складають так звані явища гістерезису, коли в наявності є декілька різних фаз, що реалізуються в залежності від початкових станів. Скажімо, при «холодному» старті (спіни в початковій конфігурації спрямовані в один бік) ми прийдемо до впорядко-
22
ваного стану, а при «гарячому» (спіни в початковій конфігурації спрямовані випадковим чином) – до невпорядкованого.
Застосування алгоритму Метрополіса до ансамблю систем, які складаються з двох спінів та термостату, призводить до зміни кіль-
костей систем у можливих конфігураціях N−−, N−↓, N↓− і N↓↓ у напрямку їх рівноважних значень, що відповідають канонічному роз-
поділу: N−↓/N−−=N−↓/N↓↓=N↓−/N−−=N↓−/N↓↓=ехр(–2J).
Згідно з алгоритмом Метрополіса, імовірності переходів
р(−−→−↓)=р(−−→↓−)=р(↓↓→−↓)=р (↓↓→↓−)=ехр(–2J); (1.15) р (−↓→−−)=р (−↓→↓↓)=р (↓−→−−)=р (↓−→↓↓)=1. (1.16)
Зміна числа систем N−↓ за один Марковів крок за рахунок переходів −↓→−− та −−→−↓ складатиме
N−↓=n−↓→−−–n−−→−↓=N−−(N−↓/N−−–exp(–2J)). (1.17)
Якщо, наприклад, число систем N−↓ більше рівноважного значення (більше відношення N−↓/N−−), то частіше відбуватимуться переходи −↓→−−, тобто число систем N−↓ буде зменшуватись, доки відношення N−↓/N−− не відповідатиме Гіббсовим вагам ( N−↓=0). Якщо ж відно-
шення N−↓/N−− менше рівноважного, буде більше переходів −−→−↓, що призведе ансамбль до правильних співвідношень між кількостями систем.
Практично, при чисельному моделюванні ми зазвичай маємо один Марковів ланцюг. Зазначений ансамбль систем можна замінити досить довгими «часовими» відрізками цього ланцюга. Середнє від будь-якої величини за ансамблем таких відрізків і є середнє за Марковим часом. Цей доказ легко переноситься на будь-яку іншу систему з дискретними або безперервними ступенями вільності.
У алгоритмі термостату [9] на кожному кроці часу Маркова один зі ступенів вільності приводиться у теплову рівновагу із зовнішнім «термостатом», що має певну температуру Т, при цьому враховується взаємодія з іншими (фіксованими на даному кроці) ступенями вільності. Внаслідок багаторазового повторення такої процедури для всіх ступенів вільності за досить довгий час Маркова настає повна термодинамічна рівновага всієї системи з термостатом. Наприклад, в моделі Ізінга на кожному кроці положення чергового спіна обирається з імовірністю, що визначається лише температурою термостата і конфігурацією сусідніх спінів. Нормовані імовірності станів з енер-
23

гіями Еi+ і Еi– в i-й конфігурації оточуючих спінів дорівнюють відповідно:
pi+
pi−
exp(−βEi+ )
= + − , (1.18) exp(−βEi ) + exp(−βEi )
exp(−βEi− )
= + − . (1.19) exp(−βEi ) + exp(−βEi )
Знаменники у ці формули введені для того, аби сума ймовірностей pі++pі– дорівнювала одиниці.
Алгоритм термостата, так само як і алгоритм Метрополіса, швидко переводить систему в область найбільш імовірних значень у фазовому просторі. Стан системи наблизиться до рівноважного, якщо зробити порядку 100 випробувань для кожного її ступеня вільності (координати частинки). Після цього значення фізичних характеристик системи тільки флуктують, і ланцюги цих значень можна використовувати для обчислення усереднених характеристик системи.
За алгоритмом термостата або алгоритмом Метрополіса моделюють граткові системи, що містять до 107 дискретних або безперервних ступенів вільності. В багатьох наукових центрах світу використовують комп’ютерні кластери, багатопроцесорні комплекси. Зараз найбільш ефективним вважається моделювання на графічних процесорах, які складаються з досить великої кількості вкладених процесорів.
1.2.4. Граничні умови
Доки комп'ютерна техніка дозволяла моделювати системи лише малого об'єму (порядку 102-104 частинок), неминуче виникало питання про екстраполяцію на великі системи, що призводило до необхідності розгляду питання про граничні умови. Вибір граничних умов залежить від поставленої задачі. У разі застосування вільних граничних умов приграничні атоми утворюють вільну поверхню, що контактує з вакуумом; атоми можуть переміщуватись так само, як і всередині об’єму. У разі застосування жорстких граничних умов координати граничних атомів фіксовані. Припускається, що досить велика кількість рухливих атомних шарів компенсує вплив таких умов на результати моделювання.
У більшості досліджень за МК методом для збереження постійного числа частинок i розгляду області моделювання як елемента нескінченного об’єму на неї накладалися періодичні граничні умо-
24

ви. В цьому разі за кожним із напрямків у гратці перший і останній ступені вільності в даному ряді взаємодіють так, неначе вони сусіди. Таким чином, за кожним з напрямків гратка замикається в кільце (вона утворює тор відповідної розмірності).
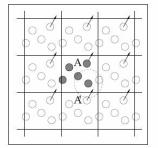
Як можна бачити з рис. 1.3,а, атом А0 взаємодіє не тільки з атомом А з основної області, що розглядається, а й з уявним атомом А’, зміщеним відносно А на період ідентичності. Якщо ж атом А знаходиться в лівій нижній чверті, його образ А’ визначається зсувом на період за двома напрямками (рис. 1.3,б)
Рис. 1.3. Схема взаємодій приграничного атома.
У разі, коли частинки можуть переміщуватися, зазвичай використовують наступні періодичні граничні умови. Якщо в результаті зміни координат частинка А виходить за межі кубічної області через певну грань, то через протилежну грань буде введена частинка, А’, котра зветься "образом". При цьому введена частинка буде займати в об'ємі куба місцеположення, координати якого обчислюють за формулою:
x |
|
− L, |
x |
|
> L; |
(1.20) |
x′A = |
A |
|
|
A |
|
|
xA + L, |
xA < 0, |
|
||||
де і - декартовий індекс (і=1,2,3), хА – координата частинки після зсуву, х'А – координата "образу", L – лінійний розмір комірки. Таким же чином перетворюються інші координати. У результаті замість частинки вводиться її образ з тим самим номером. Загальна кількість частинок в системі при цьому не змінюється. Введення "образу" продемонстровано на рис. 1.4. Таким чином, кожна частинка після зсуву опиняється в основній кубічній комірці із частинок, оточеній комірками з "образів". Це компенсує вплив границь при обчисленні енергії взаємодії.
25
Перевагою періодичних граничних умов є те, що всі ступені вільності стають рівноправними. Область МК моделювання "вбудовується" у середовище, при цьому усуваються небажані поверхні i створюється квазінескінченний об’єм, який дозволяє розглядати основну комірку як елемент нескінченної системи. Проте періодичні крайові умови можуть наводити (і дійсно наводять) значний додатковий порядок у конденсованих системах. Періодичні граничні умови є доцільними, якщо в певному напрямку модельної схеми можна виявити період повної ідентичності, що дозволяє імітувати нескінченну протяжність речовини (кристала) в розглянутому напрямку. У разі багатофазних систем їх застосовують у напрямках, де немає міжфазних меж. Треба також мати на увазі, що поява кристалічного зародку поблизу періодичної межі миттєво ініціює його ріст з протилежного боку.
Рис. 1.4. Пересування частинки та її образів при застосуванні
періодичних граничних умов,
у сферу обрізання включаються як частинки з основної комірки, так і їхні образи.
Наразі, коли персональні комп’ютери дозволяють моделювати системи у сотні тисяч атомів, є змога зовсім не застосовувати періодичні умови, а розглядати системи з вільними поверхнями, або застосовувати періодичні умови тільки за одним або двома напрямками. Системи з відкритою поверхнею краще відповідають реальним системам у разі вивчення фазових переходів. При досить великих розмірах системи вплив меж на значення екстенсивних (тобто тих, що залежать від розміру системи) фізичних величин незначний, оскільки частка поверхневих атомів мала. У багатьох випадках вибір правильних крайових умов може стати вирішальним для усього експерименту.
26
1.2.5. Вибір потенціалу взаємодії атомів
Питання ви6оpу потенціалу взаємодії є суттєвим при моделюванні атомної структури речовини. Вибір потенціалу має залежати від типу зв'язків в конкретній речовині, фізичних уявлень про природу взаємодії частинок у ній.
Потенціали обчислюють у різні способи. Теоретичні потенціали, розраховані за законами квантової механіки, враховують розподіли густини зарядів (електронної густини) [10]. Емпіричні потенціали розраховують на основі експериментальних даних щодо дифракційних досліджень структури речовини (структурного фактору, парної функції радіального розподілу атомів) [11]. Ті та інші потенціали корегують, аби результати моделювання відображали фізичні властивості відповідної речовини, такі як: пружні сталі, температурну залежність коефіцієнту розширення, температуру плавлення, теплоємність. Задовольнити всі ці вимоги вдається далеко не завжди. І твердження про те, що потенціал відноситься саме до цієї речовини, не завжди є обґрунтованим. Особливу цінність мають дослідження, які виражають загальні закономірності тих чи інших фізичних процесів, так що неточності потенціалів не змінюють характер процесів, що відбуваються.
Залежно від природи речовини, що моделюється, використовують парні, тричастинкові та багаточастинкові потенціали. У випадку останніх потенціальна енергія системи є функцією не тільки відстаней до атомів, що оточують розглядуваний атом, а й від їх координат (кутів, які вони утворюють з розглядуваним атомом). У разі парних потенціалів враховуються тільки відстані між атомами. Результат їх застосування відповідає дійсності, якщо енергія взаємодії двох частинок не залежить від розташування інших частинок. Така ситуація має місце для простих рідин та аморфних речовин, для різних станів металів. Тричастинкові потенціали застосовують до описів систем з ковалентними зв’язками. Багаточастинкові потенціали корисні для описів більш складних систем, наприклад, твердих тіл з дефектами структури. У найпростішому випадку взаємодія частинок може бути парною. При цьому енергія взаємодії системи:
U (r1 , r2 ..., rN ) = ∑ϕ(rij ) , |
(1.21) |
i< j |
|
де ri – координата i-ї частинки, rij – відстань між i-ю та j-ю частинками.
27

У принципі, функцію ϕ(r) для електронейтральних атомів можливо побудувати шляхом детального розрахунку, що базується на основних законах квантової механіки. Такий розрахунок є дуже
складним і, окрім того, зазвичай буває цілком достатньо в якості ϕ(r) вибрати просту феноменологічну формулу.
Найбільш важливими особливостями ϕ(r) для простих рідин є сильне відштовхування для малих r і слабке притягання на великих відстанях. Відштовхування на малих r зумовлене правилом заборони. Інше кажучи, якщо електронні хмари двох атомів перекриваються, деякі електрони повинні збільшити свою кінетичну енергію, аби перебувати в різних квантових станах. Слабке притягання при великих r обумовлено головним чином взаємною поляризацією атомів; результуюча сила притягання зветься силою Ван-дер-Ваальса.
Найбільш поширений з безперервних потенціалів, що застосовують до систем з електронейтральних або молекул – двопараметричний потенціал Леннарда-Джонса:
ϕ ( ) = ε r0
LJ r
r
12 |
r0 |
|
6 |
|
||
|
− 2 |
|
|
|
або |
|
r |
||||||
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
12 |
|
|
d |
||
ϕLJ |
(r) = 4ε |
|
|
|
|||
|
r |
||
|
|
|
|
−d 6
, (1.22)
r
де величина ε=V(r=r0=21/6 d) – глибина потенціальної ями, d – координата нуля потенціалу, тобто φLJ(d)=0.
Рис. 1.5. Графік потенціалу Леннарда-Джонса
Графік потенціалу Леннарда-Джонса показаний на рис. 1.5. Залежність r-6 у формулі (1.22) одержана теоретично, а залежність r –12 вибрана тільки з міркувань зручності. Відмітимо, що даний потенціал є короткодіючим, тобто практично V(r)=0 для r > 2.5 d.
Більш загальними є потенціал Мі:
28
ϕMi |
(r) = |
ε |
|
r0 |
m |
|
|
n |
|
|
|||
|
|
|||||
|
|
m − n |
r |
|
||
|
|
|
|
|
|
|
та трипараметричний потенціал Морзе:
r0 |
n |
|
||
− m |
|
|
|
(1.23) |
|
||||
r |
|
|
|
|
|
|
|
|
|
|
|
|
r |
|
|
|
|
r |
|
|
|
|
|
|
|
|
|
|
|
||
ϕM (r) = ε exp |
− 2α r |
−1 |
|
− 2 exp |
− α r |
−1 |
. (1.24) |
|||
|
|
|
0 |
|
|
|
|
0 |
|
|
|
|
|
|
|
|
|
|
|||
Усі ці потенціали мають круту гілку відштовхування, і прямують до нуля на великих відстанях. Для зменшення часу обчислень вво-
диться обрив потенціалу на деякій відстані rс, тобто взаємодія з віддаленими частинками не враховується [10].
З дифракційних експериментів із розсіювання рентгенівських променів або нейтронів можна отримати такі важливі структурні характеристики як структурні фактори і парні кореляційні функції. Втім, для отримання більш повної картини структури речовини необхідно побудувати відповідні атомні моделі, які наочніше зображують просторове розташування частинок системи. Для побудови моделі необхідно застосовувати відповідний для даної речовини потенціал. Емпіричні потенціали підбирають таким чином, щоб їх застосування призводило до структур, узгоджених з експериментальними даними за парними функціями радіального розподілу атомів (ПФРРА) та структурним фактором [11].
Емпіричний міжатомний потенціал – один із найбільш простих i доступних методів дослідження динамічних i структурних властивостей речовин. Такий потенціал описує міжатомні взаємодії речовини i містить певну кількість параметрів, які корегуються. Ці параметри пристосовані до експериментальних даних, i результати обчислювальних експериментів настільки точні, наскільки це можливо.
У разі ковалентних кристалів, напівпровідників, що мають близько 4-х зв’язків на атом, застосовують потенціали, які враховують кути розглядуваного атома з кожною парою його найближчих сусідів. Звісно, що для цього треба будувати, щонайменше, тричастинкові потенціали. При цьому запис для енергії взаємодії системи має вигляд:
U = U0 + C1∑∑ u(ri ,rj )+ C2 ∑∑∑ v(ri ,rj ,rk ).. (1.25)
i j i j k
Сумування проводиться за найближчими сусідами центральної частинки. Тут U0 – енергія ідеального кристала, C1, C2 – параметри, за допомогою яких досягається найкраща відповідність моделі з експе-
29
риментом; u(ri , rj ), v(ri , rj , rk ) – функції для опису зміни енергії че-
рез розтягнення (стиснення) зв’язків і змін валентних кутів, відповідно. Найбільш простий вигляд цих функцій запропонував Кітінг [12]
|
u(r , r |
) = |
|
r − r |
|
2 |
− d 2 , |
(1.26) |
|||
|
|
|
|||||||||
|
1 |
2 |
|
|
1 |
2 |
|
|
|
0 |
|
де r1, r2 – |
радіус-вектори сусідніх іонів, d0 – |
рівноважна довжина |
|||||||||
зв’язку. |
v(ri ,rj ,rk )= (cosθijk |
− cosθ0ijk )2 , |
|
||||||||
|
(1.27) |
||||||||||
де θijk – |
кут між зв’язками i-j і j-k, аθ0ijk |
– |
відповідний рівноважний |
||||||||
кут.
Потенціал Кітінга успішно застосовується для дослідження енергії пружної деформації в алмазоподібних кристалах.
Емпіричний міжатомний потенціал Стілінджера-Вебера [13] також широко використовується для досліджень структурних i динамічних властивостей кремнію. Енергія взаємодії в системі має вигляд:
U = |
1 |
∑ϕ(rij ) + |
∑ g(rij )g(rik ) cosθijk + |
1 |
2 |
, |
(1.28) |
|
|
|
|||||||
|
2 i, j |
i< j<k |
|
3 |
|
|
||
де g(rik ) – це спадаюча функція зі сферою обрізання між першою та
другою сферами найближчих сусідів.
Для вивчення коливального спектру і динаміки релаксації дефектів у кристалічних гратках напівпровідників застосовують потенціал Терзоффа [14]:
|
|
|
|
|
|
U = ∑Uij;k , |
(1.29) |
|
|
|
|
|
|
i< j;k |
|
де Uij;k = fc (rij )(aij fR (rij ) + bij fA (rij )) ; fc (rij ) – |
функція обрізання |
||||||
f |
R |
(r )= Ae−λ1r |
і |
f |
A |
(r )= −Be−λ2r – відповідно, |
складові відштовху- |
|
ij |
|
|
ij |
|
||
вання і притягання потенціалу (індекси R і A від англ. “repulsive” і
“attractive” ); bij = b(rij, rjk, θijk).
Зв’язок i-j послаблюється через присутність інших зв’язків i-k, котрі також має атом i. Ступінь ослаблення визначається розташуванням інших зв’язків. Для побудови реалістичної моделі кутові складові виявляються необхідними. Звісно, через чималу кількість параметрів, їх підбір, а, отже, і побудова реалістичного потенціалу є нелегким завданням.
30