

Ovru-all
.pdfr& = V |
1/3 |
s& + (1/3)V |
−2/3 & |
|
Vs , |
яке може бути отриманим з рівнянь (7.34). Метод не реалізується точно у схемах розрахунків, оскільки величина s& не розраховується на кожному ітераційному кроці за часом t. Параметр Q підбирається стосовно до розглядуваної системи, оскільки мале значення Q призводить до швидких коливань об’єму розрахункової комірки, що унеможливлює утримання тиску постійним. Велика “ маса” призводить до повільних змін об’єму, а нескінченна – до звичайного МД моделювання. В роботах [51,52] запропоноване значення величини
Qs4 /m = 0.0027 в системі атомів з потенціалом Леннарда-Джонса,
де s – параметр потенціалу Леннарда-Джонса. Ізобарно-ізоентальпійний ансамбль майже не застосовують. Най-
частіше метод постійного тиску комбінують з одним із методів забезпечення постійної температури – NPT ансамбль. Для того щоб це зробити, Берендсен [46] модифікував Лагранжіан, що призвело до появи додаткових рівнянь руху, окрім (7.36), які забезпечують утримання тиску постійним:
dPM /dt = (P − PM )/tP , |
(7.37,а) |
||||
r& = χ1/3r , |
|
(7.37,б) |
|||
χ = 1 − β |
|
δt |
(P − P), |
(7.37,с) |
|
|
|
||||
|
T tP |
M |
|
||
де P – бажаний тиск, tp – константа часу. На кожному кроці розраховується об'єм комірки, який пов’язаний з фактором c, а координати центру мас пов’язані з величиною c1/3, bT – ізотермічна стисливість; dt – крок за часом. Точно знати bT необов’язково, оскільки цей фактор може бути включеним у часову константу tp. Берендсен [46] знайшов, що використання значень tp, з інтервалу від 0.01×10-12c до
0.1×10-12c, призводить до задовільних результатів під час моделювання води. Метод істотно не впливає на динамічні траєкторії й легко реалізується у програмах. Але тип ансамблю, що застосовується, важко визначити.
В роботі [48] було запропоновано базові рівняння для забезпечення NPT ансамблю в дусі методики Андерсена [45]
& |
1/3 |
, |
(7.38,а) |
s = p/mV |
|
||
& |
|
|
(7.38,б) |
p = f - (c + x)p , |
|||
|
|
|
231 |
|
|
|
& |
|
(7.38,в) |
|
|
|
|
|
ξ = f kB (Tм − Т)/Q , |
||
|
|
|
& |
/3V , |
(7.38,г) |
|
|
|
|
|
χ = V |
||
|
|
|
& |
2 |
(7.38,д) |
|
|
|
|
|
|||
|
|
|
|
χ = (PM − P)V /tp kB T , |
||
де Tм = ∑ |
|
pi |
|
2 /(mf kB ) – миттєва температура, tp – |
час релаксації |
|
|
|
|||||
i |
|
|
|
|
||
для встановлення тиску, Pм – |
миттєве значення тиску. |
|
||||
7.3.2. Досягнення рівноважного стану та вимірювання макроскопічних величин
Під час моделювання за методом МД процес встановлення рівноваги часто займає левову частину від загального часу розрахунку. Як правило, в початкових умовах зручніше за все вибирати «рівноважну» конфігурацію з якого-небудь колишнього розрахунку, яка відповідає температурі і густині, близькій до тих параметрів, що вимагаються. Молекулярна динаміка розвиває молекулярну конфігурацію кінцевого розміру, змінюючи її в часі крок за кроком. Є межі для типових часових рамок і масштабів довжини, які можуть бути досліджені.
Виконувані при моделюванні 103÷105 кроків за часом (≤10– 15с) алгоритму MД, звичайно відповідають декільком наносекундам реального часу. Необхідно перевірити, чи була в системі досягнута рівновага перш, ніж обчислювати середні значення величин. Крім того, потрібно піддати результати моделювання статистичному аналізу, зробити реалістичну оцінку похибок. Як довго потрібно проводити розрахунок? Це залежить від системи й фізичних властивостей, що становлять інтерес.
Припустимо, що нас цікавить змінна a, визначена так, що <a>=(1/N) ∑ ai =Const в рівноважному стані (наприклад, aі – це
i
швидкість і-го атому). Введемо часову функцію кореляцій значень a
– ∑ ai (t0) ai (t0+t), що зв'язує значення, обчислені через проміжки
i
часу t. Позначимо її як <a(t0)a(t0+t)>. Якщо стан системи рівноважний, ця функція не залежить від вибору початку відліку часу й тому можна її записувати у вигляді <a(0)a(t)>. Зрозуміло, що <a(0)a(0)>=<a2>. У разі великого часу t
232
lim a(0)a(t) |
= lim a(0) a(t) = 0 , |
(7.39) |
t→∞ |
t→∞ |
|
оскільки змінні a(0) і a(t) вже не корелюють. Отже, функція кореляції
затухає, це затухання відбувається за характерний час τa. Формально можна визначити час кореляції зі співвідношення
τ |
|
= ∫∞ |
a(0)a(t) |
dt . |
(7.40) |
a |
|
||||
|
0 |
a2 |
|
|
|
|
|
|
|
|
Якщо кореляції згасають у часі за експоненціальним законом, час τa може бути визначений за формулою
a(0)a(t) exp(−t/τa ). |
(7.41) |
У межах характерного часу τa, має місце висока кореляція значень а. Подібне твердження може бути зроблено також про властивості, для яких можна знайти просторові середні за модельним об’ємом L3. Просторові функції кореляції визначаються через значення розглядуваної величини в точках з координатами rоі, що відповідають, наприклад, початковим координатам частинок у момент часу t=0, і значенням цієї величини у різних точках rі,, порівняно близьких до rоі. Позначатимемо такі кореляції як – < a(r0)a(r)> .
Як правило, значну частину рахункового часу при моделюванні займає формування рівноважного стану і громіздка арифметика, яка необхідна для обчислення сил й енергії. Якщо радіус дії сили достатньо малий, то є ряд способів зменшити час отримання рівноважного стану. Припустимо, наприклад, що потрібно змоделювати тривимірну систему з 8000 частинок. Можна спочатку змоделювати меншу систему з 1000 частинок і дати можливість цій малій системі прийти до рівноваги за потрібної температури. Після того, як рівновага досягнута, малу систему можна дублювати в кожному просторовому напрямі й створити необхідну систему з 8000 частинок. Після цього процедура встановлення рівноваги поновлюється. В повній системі рівновага звичайно встановлюється швидко.
Слід відмітити, що моделювання на графічних процесорах дозволяє досліджувати системи, які містять ~105÷106 атомів, а час розглядуваних процесів може складати десятки мікросекунд [53], що в деякому наближенні відповідає реальному експерименту.
233

Рівноважний макростан характеризується декількома параметрами, такими як, наприклад, абсолютна температура Т і середній тиск Р, а також об'ємом V й повною енергією E.
Кінетичне визначення температури випливає з теореми про рівнорозподіл: кожний квадратичний член, що входить у вираз для енергії класичної системи, що знаходиться в рівновазі за температури Т, має середнє значення ½kBT. Звідси можна визначити температуру Т системи в d-вимірному просторі співвідношенням
d |
NkBT = ∑ |
1 |
mvd2 , |
(7.42) |
2 |
|
|||
2 |
|
|
||
в якому сума береться за всіма N частинками системи й d компонентами швидкості. Дужки < > означають усереднення за часом. Вираз (7.42) є прикладом зв'язку макроскопічної величини, в даному випадку температури, із середнім за часом по траєкторіям частинок (миттєва кінетична енергія системи флуктуює). Співвідношення (7.42) справедливе у такому вигляді в тому випадку, якщо швидкість руху центру мас системи дорівнює нулю.
Ще однією тепловою характеристикою системи є теплоємність при постійному об'ємі СV=(∂Е/∂Т)V, СV – це міра кількості тепла, необхідного для зміни температури на 1 К. Оскільки теплоємність залежить від розмірів системи, зручно визначити питому теплоємність на частинку, а саме cV =СV/N. Легше за все отримати cV шляхом знаходження середньої потенціальної енергії та середньої повної енергії при близьких температурах Т й Т + Т. Температурна залежність cV зумовлена температурною залежністю потенціальної і кінетичної енергії, остання прямо пропорційна температурі ~(d/2)kBT.
Метод визначення тиску на основі теореми про віріал сили описано у підрозділі 7.1.4. У лабораторній роботі № 13 (у допоміжному посібнику) тиск визначається через імпульси молекул, що перетина-
ють межі основної комірки. Розглянемо елемент поверхні A і припустимо, що середня кількість руху, що перетинає в одиницю часу поверхню зліва направо, буде К+, а К– – середня кількість руху, що перетинає поверхню справа наліво. Тоді середня сила F рівна F = 0.5(К+ + |К– |), а середній тиск визначається виразом
P=Fn / A, |
(7.43) |
234

де Fn обозначає компоненту сили, нормальну до елемента поверхні. У двовимірному випадку тиск дорівнює густині потоку імпульсу через одиничний відрізок, а не через одиничну поверхню.
7.3.3. Властивості перенесення
Розглянемо властивості атомів, які пов’язані з їхньою рухливістю. Уявімо, що ми слідкуємо за траєкторією певної частинки. Нехай у довільно обраний момент часу t1 її місцеположення визначається вектором r1. Визначимо зміщення частинки у деякий наступний момент часу t2. Відомо, що якщо сумарна сила, діюча на частинку Fi, дорівнює нулю, то її зміщення росте з часом лінійно. Однак у конденсованому стані кожна частинка зазнає багато зіткнень, і в середньому її сумарний зсув практично дорівнює нулю. Інтерес становить величина середнього квадрата зміщення, що визначається формулою
|
= |
1 |
N |
[r |
(t) − r (0)]2 |
|
|
|
r 2 |
∑ |
, |
(7.44) |
|||||
|
||||||||
t |
|
N i=1 |
i |
i |
|
|
||
де  ...
... – дужки статистичного усереднення за всіма частинкам сис-
– дужки статистичного усереднення за всіма частинкам сис-
теми. Оскільки система перебуває в рівновазі, початок відліку часу є довільним і середнє в (7.44) залежить тільки від інтервалу часу t. Відомо, що залежність від t функції < r2(t)> дозволяє розраховувати коефіцієнти дифузії. Коефіцієнти дифузії D за деякої температури Т можливо розраховувати за нахилом залежності середньоквадратичного зміщення < r2(t)> частинок від часу t
< r2(t)>=6Dt+C, |
(7.45) |
де С – константа, яка характеризує відхилення динаміки системи від марковської (для марковських процесів С=0).
Для характеристики динамічних властивостей частинок розрахо-
вують автокореляційну функцію швидкості (АФШ) [54]. vi(t) – це випадкова величина швидкості частинки в момент часу t за умови, що в початковий момент часу її швидкість дорівнювала vi(0). Середнє значення <v(t)>=0. Якщо сумарна сила, яка діє на і-ту частинку, дорівнює нулю, то швидкість останньої буде постійною. Однак за рахунок взаємодії з іншими частинками швидкість частинки буде змінюватися, й можна очікувати, що через певний час її швидкість не буде помітно корелювати з початковою швидкістю. За визначенням, АФШ – це записана нижче функція Z(t):
235
Z(t) = |
vi (0)vi (t) |
|
vi (0)vi (0) , |
(7.46) |
де vі(0) й vi(t) – вектори швидкостей в початковий момент часу й у моменти часу t,  ...
... – дужки статистичного усереднення за всіма
– дужки статистичного усереднення за всіма
частинкам системи.
Динаміку атомів можна характеризувати спектром коливань атомів g(ω), отриманим з Фур’є перетворення АФШ
|
1 |
∞ |
|
g(ω) = |
|
∫ Z(t)exp(−iωt)dt . |
(7.47) |
|
|||
|
π 0 |
|
|
Для графічного відображення відокремлюють дійсну частину з рівняння (7.47)
|
2 |
∞ |
|
g(ω) = |
|
∫ Z(t)cos(ωt)dt . |
(7.48) |
|
|||
|
π 0 |
|
|
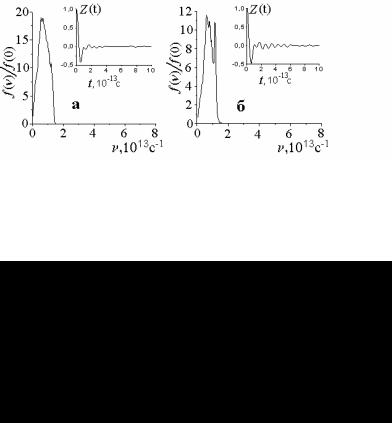
Рис. 7.16. Автокореляційні функції швидкості та їхні спектри в системах чистого заліза з нанокристалами різного розміру [105], Т=300 К
а – малі нанокристали (@350 атомів), b – великі нанокристали (³2000 атомів)
За виглядом залежності функції Z ( t ) , а саме за її затуханням, можна судити про динамічну релаксацію в матеріалах. На графіках рис. 7.16,а,б видно, що час динамічної релаксації в матеріалах з нанокристалами різного розміру відрізняється. У випадку зразків з нанокристалами малого розміру (рис. 7.16,а) й аморфних зразків осциляційний характер АФШ й їх спектри майже не відрізняються. Для атомів у цих зразках характерні неузгоджені коливання. У випадку зразків з нанокристалами великого розміру осциляції АФШ затухають повільніше, а у спектрах спостерігається чітке роздвоєння максимуму (рис. 7.16.,б). Нижче, на рис. 7.30, наведені інші данні, які
236

також підтверджують, що спектральні характеристики атомів різні в зразках з нанокристаллами різного розміру.
Автокореляційна функція швидкості пов’язана з дифузійною рухливістю атомів системи. Формула для розрахунку коефіцієнта дифузії D має наступний вигляд:
|
1 |
∞ |
N |
|
|
D = |
∫0 |
∑ vi (0)vi (t) dt , |
(7.49) |
||
3N |
|||||
|
i=1 |
|
де N – кількість атомів в системі, vi(0) – швидкість і-го атому в початковий момент часу, vi(t) – швидкість і-го атому в момент часу t. Фактично, цю формулу можна застосовувати для рідкого стану, для якого коефіцієнти дифузії досить великі.
Важлива інформація щодо збереження ближнього порядку в просторовому розподілі атомів аморфних та рідких матеріалів може бути отримана з розрахунку залежної від часу просторово-часової ко-
реляційної функції Ван-Хова [55]. Відомо [56], що розраховані під час моделюванні за методом МД кореляційні функцій Ван-Хова можна порівняти з експериментальними даними, а саме з інтенсивністю розсіяння повільних нейтронів речовиною.
Спрощений варіант розрахунку однією із функцій Ван-Хова Gd(r,t) запропоновано Рахманом [56]:
Gd (r, t) = Ω0 [n(r, t) / 4πr2 r], |
(7.50) |
де n(r,t) – число частинок в момент часу t, розташованих в інтервалі відстаней від r до r+ r, (r – це відстані від положення частинок відліку у момент часу t=0), Ω0 – об’єм, який припадає на одну частинку.
У момент часу t=0 функція Gd(r,t) співпадає з g(r) – рівноважною парною радіальною функцією розподілу частинок. Часовий розвиток функції Gd(r,t) відповідає релаксації з часом початкового розподілу g(r), тобто втратою з часом початкових просторових кореляцій, що пов’язано із зміщеннями частинок.
Методом МД може бути отримана ще одна функція Gs(r,t), яка дає густину імовірності знаходження частинки в точці r в момент часу t, якщо при t = 0 вона знаходилась в початку координат. Згідно
з [57],
|
(r,t) = |
1 |
N |
δ[r−|r |
(t) − r (0)|] , |
|
|
G |
∑ |
(7.51) |
|||||
|
|||||||
s |
|
N |
i=1 |
i |
i |
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
237 |
де |ri(t)-ri(0)| – модулі зміщення атомів, а функція δ[r−|ri (t) − ri (0)|]дорівнює одиниці у випадках, коли |ri(t)-ri(0)| належить інтервалу від r до r+ r, інакше ця функція дорівнює нулю.
Зрозуміло, що залежність середньоквадратичного зміщення частинок від часу визначається характером функції Gs (r,t):
r2 = ∫ r2Gs (r,t)dr . |
(7.52) |
Вважають, що функція Gs(r,t) частково підпадає під закон розподілу Гауса, й вивчають, якою мірою ця функція відхиляється від розподілу Гауса на всьому інтервалі значень (r,t). Більше інформації про функції Ван-Хова можна знайти в оглядах [57,58].
7.3.4. Топологічний аналіз змодельованих атомних конфігурацій. Метод Вороного-Делоне
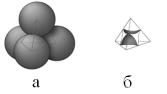
Сучасне дослідження структури потребує математичного підходу для вивчення взаємного розташування молекул у просторі, не опираючись ні на хімічні зв'язки, як при вивченні структури окремих молекул, ні на принципи й властивості трансляційної симетрії, які розроблені в кристалографії. Подібний підхід вже давно був розроблений математиками [59,60]. Одним з головних його творців був Г.Ф.Воронов. Б.Н. Делоне пояснив суть робіт Вороного й узагальнив основні теореми [60]. Бернал вперше застосував метод Вороного до дослідження структури рідини [61]. Вихідною геометричною побудовою, на якій базується метод, є так званий багатогранник Вороного – область простору, найближча до даного атома. Для його визначення посередині відрізків, проведених до найближчих атомів, будуються перпендикулярні до них площини (див. рис. 7.17,a). Фактично це комірка Вігнера-Зейтца. Форма багатогранників Вороного залежить від конкретного розташування сусідів навколо даного атома. Побудова багатогранників Вороного дозволяє перейти від переліку (файлу) координат атомів до геометричних образів. На рис. 7.18 показані багатогранники Вороного деяких тривимірних упакувань куль. Для простих кристалічних структур існує свій єдиний багатогранник, що відбиває найближче оточення, однакове для всіх атомів даного кристалу. У неупорядкованій фазі багатогранники Вороного, природньо розрізняються для різних атомів, але вони мають характерні риси, які відповідають структурі даної системи У сучасних роботах з моделювання показано, що в аморфних фазах досить велика доля кластерів, утворених ікосаедрами (20-гранник, утворений з трикутникових пірамід, рис. 7.18,д), для яких
238

багатогранник Вороного показано на рис. 7.18,г). Ікосаедри звичайно є центрами квазікристалів, розміри яких обмежені в силу відсутності трансляційної симетрії.
Рис. 7.17. Ілюстрація побудови багатогранників Вороного (а),
двовимірна ілюстрація розбиття Вороного (б).
Рис. 7.18. Багатогранники Вороного для деяких систем:
проста кубічна гратка (а); гранецентрована кубічна гратка (б); гратка алмазу (в); ікосаедрична конфігурація (г) (ікосаедр показаний схемою (д), його основні осі симетрії 5 порядку, атом у центрі має 12 найближчих сусідів); довільне оточення атома (е).
Робота з багатогранниками Вороного, це частина методу Вороно- го-Делоне. Головні можливості методу базуються на використанні не окремих багатогранників, а цілком всієї мозаїки багатогранників Вороного. Важливим моментом є те, що багатогранники Вороного заповнюють простір без накладень і щілин, тобто здійснюють, як кажуть математики, розбиття простору. На рис 7.17,б показана двовимірна ілюстрація такого розбиття. Пунктирними лініями показаний симплекс Делоне. Вершинами симплекса Делоне є центри атомів. Симплекси Делоне системи заповнюють простір без накладень і щілин, роблячи розбиття Делоне. Сфера, описана навколо симплекса Делоне порожня, у ній немає центрів інших атомів. Одночасно можна всю систему атомів розбити на групи атомів – симплекси Делоне. У двовимірному просторі це трійки атомів, у тривимірному – четвірки (рис 7.19). Центри атомів визначають вершини багатогранника, найпростішого в просторі даної розмірності (у математиці такий багатогранник називається симплексом). На площині – це трикутник, у
239
просторі – тетраедр. Їхня форма визначається конкретним розташуванням атомів і може служити характеристикою структури системи.
Рис.7.19. Ілюстрація симплекса Делоне в
3-вимірному просторі;
а – симпліцільна конфігурація атомів; б – порожній простір між атомами,
що визначає елементарну (симпліцільну) пору.
Крім вивчення структурних характеристик, розбивка ВороногоДелоне дозволяє приступити до вивчення порожнеч між атомами. Цей аспект досліджень є навіть важливішим, ніж аналіз закономірностей у розташуванні атомів самих по собі. Багато важливих фізичних процесів, наприклад, дифузія домішок, протікання дрібних атомів через гранульовані пористі матеріали зв'язані напряму саме з міжатомними порожнечами. Симплекс Делоне визначає відносно велике вільне місце між атомами, куди ведуть, як правило, більше вузькі порожнечі (рис. 7.18 та 7.19). З геометричної точки зору кожен симплекс Делоне визначає елементарну порожнину в системі атомів.
Для невпорядкованих і термічно збурених систем симплекси Делоне є викривленими тетраедрами. Використовуючи кількісну міру форми симплексу, автори [62] виділяли ті атомні конфігурації, які ближче до досконалого тетраедра. Саме такі симплекси характерні для щільної кристалічної структури. Вони зустрічаються і в рідкій фазі, проте характер їх взаємного розташування принципово відрізняється від кристалічного. Питання щодо аналізу структури модельованих систем методом Вороного-Делоне докладно розглянуті в книзі [63].
7.4.Проблема вибору потенціалів
7.4.1.Потенціали міжатомної взаємодії в металах
При побудові моделей речовини методами МД і Монте-Карло виникає необхідність застосування потенціалів міжатомної взаємодії. У підрозділі 1.2.5 були розглянуті деякі потенціали міжатомної взаємодії, які застосовують для моделювання, кристалів інертних газів (парний потенціал Леннарда-Джонса) і напівпровідникових речовин Si, C, Ge (трьохчастинкові потенціали Стілінджера-Вебера, Терзоффа).
240