

Ovru-all
.pdfРозписавши суми геометричних прогресій, прийдемо до рівності
Na = N |
|
|
|
kx |
|
|
|
|
, |
|
|
|
|
|
|
|
|||||
(1 |
− x)(1 − x + kx) |
|
|
|
|
|
|
|
|||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||
|
|
|
|
|
|
|
|
|
|
= βP x = βτ1 P, kx = βτ P . |
|
||||||||||
ν = |
nV |
|
= |
|
|
P |
|
||||||||||||||
|
|
|
|
|
|
||||||||||||||||
4 |
|
|
|
|
|
2πμkT |
|
|
|
|
N |
|
N |
|
|||||||
Перепишемо останню рівність у вигляді: |
|
|
|
|
|||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
Na |
= Q = |
|
kx |
|
|
, |
(5.3) |
||
|
|
|
|
|
|
|
|
|
|
|
|
N |
(1 − x)(1 − x + kx) |
||||||||
якщо x<<1, то Q = |
|
kx |
|
– маємо рівняння Ленгмюра; |
|
||||||||||||||||
|
1 + kx |
|
|||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
1 |
|
|
|
|||
якщо x ≈ 0.5 -1.0, kx (1–x+kx), |
то Q ≈ |
|
. |
|
|
||||||||||||||||
|
|
|
|
||||||||||||||||||
|
|
|
|
||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
1 |
− x |
|
|||
Отже, рівняння БЕТ дозволяє отримати різні типи ізотерм в залежності від параметру, значення якого обумовлено фізичними факторами. Але все різноманіття ізотерм у рамках спрощеної моделі пояснити не можна.
Величина |
N |
= q |
має розмірність тиску, x = |
P |
. Якщо при зрос- |
|
|||||
|
βτ1 |
|
q |
||
танні Р досягає точку P=q, то x = 1 ; при цьому, NA → ∞ , тобто від-
бувається конденсація пари в рідину, a q має зміст тиску насиченої пари P0 для даної речовини за певної температури. Для тисків мен-
ших ніж q, x = P <1, можна розраховувати кількість адсорбованих q
молекул за наступною формулою:
Na = |
|
NkP |
|
, |
(5.4) |
|
|
|
|
|
|||
|
|
P |
|
|
||
|
(q − P) |
1 + (k − 1) |
|
|
||
|
|
|
||||
|
|
|
q |
|
|
|
де N – кількість адсорбуючих «центрів». Позначивши об’єм осаджених молекул як V = NaΩ , а об’єм моношару як Vm = NΩ , прийдемо до виразу для осадженого об’єму
V = |
|
kPVm |
|
, |
(5.5) |
||
|
(P − P) |
1+ (k − 1) |
P |
|
|
||
|
|
|
|||||
0 |
|
|
P0 |
|
|||
|
|
|
|
|
|
||
|
|
|
|
|
|
|
141 |
Осаджений об’єм можна знаходити експериментально з вимірювання добутку PV газу до і після його контакту з поверхнею. Перепишемо (5.5) у вигляді
P |
|
1 |
|
k - 1 |
|
P |
. |
V (P - P) |
= kV |
+ |
kV |
× P |
|||
0 |
|
m |
|
m |
0 |
|
|
Якщо побудувати графік залежності величини лівої частини рівності від Р/Р0, то можна знайти величину Vм, а ,отже, і площу речовини, що використовується як адсорбент. Такий спосіб визначення площі використовується й досі.
5.2.Адсорбційне рівняння Гіббса
5.2.1.Фізична межа розділу фаз
Об'єктом вивчення фізики міжфазних явищ є анізотропна і неоднорідна в одному напрямі зона безперервної зміни локальних властивостей, розташована між дотичними фазами. Цю зону Гіббс називав поверхнею розриву. Термін поверхневий шар підходить для випадку, коли щільність однієї з фаз значно менше щільності іншої, наприклад, межа рідина-газ, тверде тіло - газ. При розгляді меж тверде тіло - рідина, тверде тіло - тверде тіло переважно застосовують термін міжфазний шар.
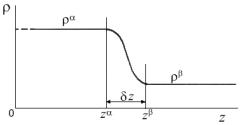
Рис. 5.4. Профіль локальної щільності на межі двох фаз.
Розглянемо, як змінюються властивості в поверхневому шарі системи рідина - пара на прикладі локальної щільності. В однорідних
фазах α і β щільність незмінна (рис. 5.4). Вісь z направлена перпендикулярно до поверхні. Залежність локальної властивості від координати в міжфазному шарі називається профілем властивості. Межі міжфазного (поверхневого) шару повинні виділяти зону істотної зміни даної властивості (див. докладніше в [1]). Взагалі кажучи, дія однієї фази розповсюджується на всю іншу фазу, тому слід говорити
142
про ефективну товщину поверхневого шару, за межами якого відхилення локальних властивостей від їх об'ємних значень стають неістотними. Проте питання про визначення меж, а отже, і ефективної товщини поверхневого шару залишається дискусійним [2].
Для рідини з ван-дер-ваальсовською взаємодією між молекулами в роботі [3] методами статистичної фізики отримана наступна асимптотична формула, що описує зміну щільності в поверхневому шарі:
ρ=ρ0+πρ02χ0(а’ρ’ – аρ0)/(σ z3), |
(5.6) |
де ρ0 і χ0 – відповідно щільність та ізотермічна стисливість однорідної рідини; a и a’ – постійні ван-дер-ваальсовської взаємодії молекул
рідини одні з одними й з молекулами іншої фази відповідно; ρ' – щільність іншої фази; z – відстань від граничної поверхні. Розрахунки по цій формулі показали, що вже на відстанях порядку декількох міжмолекулярних відстаней в рідині різниця щільності в поверхневому шарі і об'ємній фазі стає значною. Аналогічні вирази отримані для тангенціальної складової тензора тиску і показано, що тиск міняється повільніше, ніж щільність. Звідси витікає, що ефективна товщина повинна визначатися по конкретній властивості і загалом вона буде різною для різних властивостей.
При розгляді термодинаміки поверхні розділу фаз не беруть до уваги динамічний характер рівноваги поверхневого шару. Проте молекули в поверхневому шарі, особливо, якщо одна з фаз флюїдна (текуча), знаходяться в постійному русі. Якщо фази знаходяться в термодинамічній рівновазі, то існує збалансований перехід молекул з обох фаз в поверхневий шар і назад. Розрахунки показують, що при кімнатній температурі кожен см2 поверхні води щомиті приймає і віддає в парову фазу 1.2·1022 молекул. Аналогічно, унаслідок теплового руху, відбувається інтенсивний обмін молекулами між поверхневим шаром і рідкою об'ємною фазою. Поверхневі молекули твердого тіла в цьому сенсі відносно мало рухомі.
У багатокомпонентній системі профілі локальної концентрації різних компонентів можуть істотно відрізнятися. Як приклад розглянемо, як міняється локальна концентрація компонентів в трьохкомпонентній двофазній системі. Очевидно, що величина локальної концентрації залежить від нормальної до поверхні координати z (рис. 2). В стані термодинамічної рівноваги в однорідних фазах середнє число молекул будь-якого сорту в одиниці об'єму залишається постійним. У поверхневому шарі при переході від однієї фази до іншої будь-яка властивість міняє своє значення від значення цієї
властивості у одній фазі, скажемо α, до значення в іншій фазі – β.
143
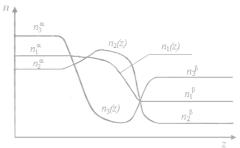
Можливі профілі локальної концентрації, показані на рис.5, можна умовно розбити на три групи:
1)монотонна функція без екстремуму з однією точкою перегину
(n1(z));
2)функція з максимумом і з двома точками перегину (n2(z));
3)функція з мінімумом і з двома точками перегину (n3(z)).
У другому випадку концентрація компоненту вища, ніж середнє значення від об'ємних концентрацій, і його називають поверхнево активним. Якщо вміст компоненту в міжфазному шарі нижче середнього значення (третій випадок), то його називають інактивним.
Рис. 5.5. Можливі профілі локальної концентрації в трикомпонентній
системі.
5.2.2. Елементарна робота деформації. Поверхневий натяг
Для двофазної багатокомпонентної відкритої системи, у разі відсутності дії зовнішніх полів внутрішня енергія
dU = TdS + dAe + ∑μidNi ,
i
де Ае – елементарна робота деформації, що здійснюється зовнішніми силами над системою.
Знайдемо вираз для роботи деформації плоского поверхневого шару товщиною Z і площею ω. Вводимо декартову систему координат з віссю z, перпендикулярною до поверхні і площиною ху, лежачою в поверхневому шарі. У загальному вигляді робота деформації записується таким чином:
dAзовн=– ∫∫∫ |
δV′∑ Pj,kε j,k , |
(5.7) |
V |
|
|
|
j,k |
|
де Pj,k і εj,k – компоненти тензора тиску і деформації відповідно.
144
У відсутності зовнішнього поля і при великій протяжності ізотропного (рідкого) шару з умов симетрії маємо:
Px,y =Py,x =Pz,x = Px,z = Pz,y =Py,z =0; Px,x = Py,y = Pτ ; Pz,z = Pn .
де Pn – нормальний тиск, величина якого в стані рівноваги постійна для всієї системи, що розглядається; Pτ – тангенціальна складова тиску, яка залежить від нормальної до поверхні координати z.
Якщо електричне і магнітне поля присутні, то треба буде ввести додаткові доданки у вираз для роботи і, відповідно, електричний і магнітний поверхневий натяг.
З урахуванням співвідношень симетрії вираз (1) можна переписати в наступному вигляді
dAзовн=– ∫∫∫V δV′[Pτ (ε x,x + ε y,y ) + Pnε z,z ]=
– ∫∫∫V δV′[Pn (ε x,x + ε y,y + ε z,z ) − (Pn − Pτ )(ε x,x + ε y,y )].
Легко зрозуміти, що εxx+εyy+εzz – це відносна зміна об'єму, а
εxx+εyy – відносна зміна площі поверхні. У останньому виразі нормальний тиск виноситься, і інтеграл за об'ємом дає його приріст dV. У другому інтегралі можна винести площу ω, оскільки Pn – Pτ залежить тільки від z. Тому
dAзовн= – РdV+γdω,
де введено позначення
Z
γ= ∫ (Pn − Pτ (z))(ε x,x + ε y,y )dz ,
0
аZ – це товщина переходного шару.
Оскільки в однорідних фазах Pn – Pτ =0, то межі інтегрування в (5) можна змінити й для двофазної системи записати:
∞ |
|
γ = ∫ (Pn − Pτ (z))(ε x,x + ε y,y )dz . |
(5.8) |
−∞
Величину γ стосовно поверхні розриву називають поверхневим натягом. Як видно з останнього виразу, поверхневий натяг обумовлений
невиконанням закону Паскаля усередині поверхні розриву (Pn ≠ Pτ ). Таким чином, поверхневий натяг плоского поверхневого шару визначається виразом (5.8), з якого видно, що ця величина інтегральна. Формулу (5.8) можна розповсюдити і на той випадок, коли на систему діють інші сили (магнітні, електричні і так далі). Відповідні доба-
145

вки до тензора тиску при цьому дадуть магнітне або електричне поверхневе натягнення.
З урахуванням (5.8) основне рівняння термодинаміки для двофазної відкритої системи можна записати в наступному вигляді:
dU = TdS − PndV + γdω + ∑μidNi . |
(5.9) |
i |
|
5.2.3. Метод Гіббса в термодинаміці поверхневих явищ
Термодинамічна теорія поверхневих явищ, створена Гіббсом більше 100 років тому, базується на таких основних поняттях, як розділяюча поверхня, система порівняння, що ідеалізується, і надлишкові екстенсивні величини.
Розглянемо багатофазну багатокомпонентну систему з поверхня-
ми “ розриву” ( міжфазного шару площею ω і товщиною Z – рис. 5.6,а), що знаходиться в стані термодинамічної рівноваги. Розгляд обмежимо плоскими поверхнями розриву. Поверхня “ розриву” характеризується градієнтом властивостей уздовж нормалі до неї і не може бути розглянута як автономна фаза, оскільки вона не може існувати без співіснуючих фаз. Застосування законів термодинаміки до цієї поверхні стає можливим, якщо ввести поняття розділяючої поверхні і термодинамічної системи порівняння, що ідеалізується.
У листі до Американської академії, написаному з приводу присудження йому премії Румфорда, Гіббс писав: "Одною з основних задач теоретичного дослідження в будь-якій області знань є встановлення такої точки зору, з якої об'єкт дослідження виявляється з найбільшою простотою". Така точка зору в теорії капілярності Гіббса, – це уявлення про розділяючу поверхню [1].
Розділяюча поверхня – це геометрична поверхня, що не має товщини (рис.5.6,б), в думках проведена між співіснуючими фазами паралельно поверхні розриву (рис.5.6,а) таким чином, що вона відтворює форму останньої. Це поняття є одним з важливих в теорії капілярності. З нею безпосередньо пов'язано поняття термодинамічної системи порівняння, що ідеалізується. У теорії капілярності Гіббса вона являє собою термодинамічну систему,
146
об'єм і форма якої такі ж, як і у реальної системи, проте властивості фаз (щільність, концентрація компонентів, ентропія і енергія в одиниці об'єму) постійні аж до розділяючої поверхні.
Таким чином, в системі порівняння між фазами відсутня неоднорідна поверхня розриву, і властивості при переході від однієї фази до іншої міняються стрибком в математичному сенсі. Для визначеності приймається, що об'єм системи порівняння залишається рівним об'- єму реальної системи, навіть у разі деформації останньої. Тобто систему порівняння вибирають таким чином, що при будь-яких змінах в
реальній системі було справедливе співвідношення Vα +Vβ =V, де Vα и Vβ – об'єми фаз в системі порівняння, величини яких залежать від положення розділяючої поверхні, V – об’єм реальної, двофазної системи.
Отже, система порівняння в теорії капілярності Гіббса - це термодинамічна система, з властивостями якої порівнюється властивості реальної системи. Об'єм і форма системи порівняння такі ж, як і у реальної системи. Проте щільність екстенсивних властивостей (концентрація компонентів, ентропія, масова щільність енергії) в ній постійна аж до вибраної розділяючої поверхні. Це об'ємні фази, що контактують, але вони позбавлені перехідного шару (рис.5.6,б). Очевидно, що ні в одній реальній системі з поверхнею розриву властивості фаз не можуть залишатися постійними аж до розділяючої ідеальної поверхні, в думках проведеної дослідником в області неоднорідності. Тому значення екстенсивних властивостей реальної системи завжди відрізнятимуться від властивостей в системі порівняння. Кількісно цю відмінність Гіббс характеризує надлишковими величинами, які представляють різниці між відповідними екстенсивними величинами в цих системах.
Розглянемо багатофазну, багатокомпонентну систему з поверхнями розриву між фазами. Кожна фаза характеризується своїм чис-
лом компонентів. Нас надалі цікавитимуть фази α і β та поверхні розриву між ними. Запишемо спочатку фундаментальне рівняння термодинаміки для багатофазної системи в цілому для області, яка містить перехідний шар (від 0 до Z) і прошарки контактуючих фаз
(рис.5.6,а):
dU = TdS − P dV + γdω + ∑ |
μ φ dN |
φ |
, |
(5.10) |
|
n |
i,φ |
i |
i |
|
|
де індекс φ приймає значення α або β ( в умовах термодинамічної
рівновагиμiα=μiβ). Диференціальне рівняння (5.10) має ту корисну властивість, що воно однаково застосовне як до всієї системи в ціло-
147
му, так і до будь-якої її частини. Запишемо подібні вирази для фаз α і β в системі порівняння.
dUa = TdSa − P dVa + ∑ |
μ a dN |
a |
; |
(5.11) |
|
n |
i |
|
i |
|
|
i,f |
|
|
|
|
|
dUb = TdSb − P dVb + ∑ μ bdN |
b . |
|
(5.12) |
||
n |
i |
i |
|
|
|
i,f |
|
|
|
|
|
Знайдемо кількісну відмінність реальної системи від уявної системи порівняння. Для цього віднімемо з рівняння (5.10) вирази (5.11) і
(5.12). З урахуванням умови d(V- Vα- Vβ) =0, яке виходить з рівності V=Vα + Vβ, маємо:
d(U − Ua − Ub ) = Td(S − Sa − Sb ) + γdω + ∑ μid(Ni − Nia − Nib ) .
i |
|
З урахуванням позначень: Uω = U–Uα– Uβ ; Sω = S–Sα–Sβ; |
|
Nω = N–Nα–Nβ, запишемо: |
|
dUw = TdSw + γdω + ∑ μi dNiw , |
(5.13) |
i |
|
де Uω, Sω і Nіω – надлишкові величини, що характеризують відмінність реальної системи від системи порівняння. Їх значення залежать від положення розділяючої поверхні.
З отриманих рівнянь видно, що якщо U є характеристичною функцією змінних S, V, Nі , то надлишкова внутрішня енергія Uω є характеристичною функцією незалежних змінних Sω, ω, Nіω . Отже, так само як і у разі об'ємних властивостей, інтенсивні властивості можна виразити через частинні похідні по екстенсивних параметрах:
T = (∂Uw /∂Sw ) |
w,Nj( j¹i) |
, γ = (∂Uw /∂ω) |
S |
ω |
,N j ( j¹i) |
, |
||||||
|
|
|
|
|
|
|
|
|||||
|
μ |
i |
= (∂Uw /∂N |
w ) |
S |
ω |
,N j ( j¹i) |
, |
|
|||
|
|
|
|
i |
|
|
|
|||||
і справедливі відповідні співвідношення Максвела.
Оскільки внутрішня енергія U і надлишкова внутрішня енергія Uω є однорідними функціями першого ступеня щодо незалежних змінних Sω, ω, Nіω, то за теоремою Ейлера маємо:
Uw = TSw + γω + ∑ μi Niw . |
(5.14) |
i |
|
Формально розписуючи повний диференціал dUω і порівнюючи отриманий вираз з (5.13), приходимо до співвідношення ГіббсаДюгема
148
SωdT + ωdγ + ∑Niω dμi = 0 . |
(5.15) |
i |
|
Можна записати вирази й інших надлишкових потенціалів:
Fω =Uω – T Sω = γω + ∑ μi Niω ,
i
Hω = Uω +P Vω = T Sω + γω + ∑ μi Niω =Uω (Vω = 0),
i
Gω = Hω –T Sω = γω + ∑ μi Niω = Fω.
i
Як бачимо, вирази для надлишкових вільних енергій Гельмгольца і Гіббса однакові, а також для ентальпії і внутрішньої енергії. Так само як і термодинамічні потенціали об'ємних фаз, надлишкові термодинамічні потенціали зв'язані одні з одними фундаментальними рівняннями і, отже, їх можна розглядати так, як ніби вони визначають деяку фазу.
Запишемо надлишкову внутрішню енергію і надлишкову вільну поверхневу енергію у розрахунку на одиницю площі:
u |
ω = Tsω + γ + ∑ μ Γ ω |
, |
|
(5.16) |
|
|
i |
i i |
|
|
|
|
= uω − Tsω |
|
|
||
σ = f |
ω = γ + ∑ μ Γ ω |
, |
(5.17) |
||
|
i i |
|
|
|
|
i
притому dσ=duω–d(Tsω)=– sωdT+γdω/ω+ ∑μidΓi .
i
Тут Гi =Niω/ω = (Ni–Niα–Niβ)/ω– величина адсорбції, тобто інтегральний надлишок i-го компоненту в порівнянні із системою порівняння у розрахунку на одиницю площі. Вільна поверхнева енергія σ рів-
на поверхневому натягненню γ у разі однокомпонентних систем, а у разі багатокомпонентної системи рівність матиме місце у випадку ∑ μ iΓi = 0 – еквімолярна поверхня. Запишемо співвідношення Гібб-
i
са-Гельмгольца для такої поверхні
uω = σ + Tsω = σ − T(∂σ /∂T)ω , |
(5.18) |
оскільки sω = –( дσ/дТ)ω=1.
З вищевикладеного випливає, що поверхневий натяг γ (формула (5.17)) не є ані внутрішньою, ані потенціальною енергією одиниці площі поверхні розриву.
149
5.2.4. Адсорбційне рівняння
Інтенсивні властивості об'ємних фаз зв'язані між собою відомим рівнянням Гіббса-Дюгема
SαdT–VαdP+ ∑μidΓi =0.
i
Аналогічне рівняння (5.15) отримане і для надлишкової внутрішньої
енергії міжфазного шару. Розділимо цю рівність на площу ω і перепишемо у вигляді.
d γ = −s |
ω dT − ∑ Γ dμ |
i |
. |
(5.19) |
|
i |
|
|
i
Приведене рівняння Гіббса-Дюгема для надлишкової внутрішньої енергії називають адсорбційним рівнянням Гіббса.
Очевидно, що
Γi= –( дγ/дμі)Т. |
(5.20) |
Для того, щоб знайти різницю якоїсь властивості у реальній системі в системі порівняння треба ураховувати об’єми α і β фаз:
x–хпр= х–хαVα–xβVβ. Оскільки V=Vα+Vβ, останню рівність можна переписати у вигляді
X– Хпр= Х– xβV– ( хα – xβ)Vα.
Отже, надлишкова величина залежить від об’єму α-фази, а значить і від положення розділяючої поверхні в системі порівняння. Для кращого розуміння теорії Гіббса необхідно з'ясувати, яка ця залежність.
5.2.5. Залежність надлишкових термодинамічних величин від положення розділяючої поверхні
На відміну від термодинамічних функцій об'ємних фаз, надлишкові термодинамічні функції залежать від положення розділяючої поверхні в системі порівняння. Проте, не дивлячись на залежність надлишкових термодинамічних функцій від положення розділяючої поверхні, загальна форма отриманих вище термодинамічних рівнянь, за винятком декількох спеціальних випадків, інваріантна щодо зміщень розділяючої поверхні.
Будь-яка надлишкова екстенсивна термодинамічна величина може бути представлена у вигляді
X ω =(Х – Хα – Хβ)/ω.
150