

Ovru-all
.pdfекспериментальними даними) значень безрозмірної ентропії плавлення ( S=ΔHПЛ/(kTПЛ)≈4.8) або параметру шорсткості Джексона
(α≈3.2), що відповідають переходу від атомно гладеньких до шорстких поверхонь. Для усунення цієї невідповідності в [33,34] прийнята модель ґраткової рідини, у якій імовірності приєднання "рідких" атомів до кристалічних кластерів залежать від числа nl їхніх зв'язків з іншими найближчими атомами у рідкій фазі:
p |
+ |
|
Δμ − 2nlηϕ |
|
− Hпл |
, |
(7.26) |
|
|
= τminν0 exp |
kT |
exp |
|
||||
|
|
|
|
|
kT |
|
|
|
де 2nlηϕ – величина додаткового бар’єру для приєднання атомів до кристала; (η – множник, що обирається таким чином, аби величина 2zηϕ дорівнювала енергії активації самодифузії в рідкій фазі); τmin – крок за часом.
Відповідно, імовірність відриву атома від кристала становить:
p– = τminνoexp(–2ns(1+η)ϕ/kT), |
(7.27) |
де ns – число s-s ("твердий"-"твердий") зв’язків атома. Енергетичний бар’єр становить f=2(1+η)ϕ на один s-s зв’язок; він складається з різниці в енергії взаємодії для атомів твердої та рідкої фази (Vll –
Vss)≈2ϕ і додаткового бар’єру 2ηϕ.
Дифузія в гратковій рідині здійснюється за вакансійним механіз-
мом, а вакансії вводились у невеликій кількості ( 3 % ). Було прийнято, що енергетичний бар’єр для стрибка атомів у вакансію залежить від числа найближчих сусідів в старому та новому положенні. Імовірність обміну місцями атома з вакансією розраховувалася згідно з рівнянням
|
= τmin |
ν |
− |
E − umin |
|
||
pстрибка |
0 exp |
|
|
, |
(7.28) |
||
|
kT |
||||||
|
|
|
|
|
|
||
де umin =nl2ηϕ – мінімальна величина енергії активації для стрибка,
2ηzϕ=QD – енергія активації самодифузії , E=∑εiα.,jβ |
niα, j,β + ∑Vi,αj,β niα,,jβ |
i, j |
i, j |
– доданок до величини енергії активації в тих випадках, коли енергія атома в новому стані вище, ніж у попередньому, (i, j – індекси ком-
понентів; α, β – індекси фаз); |
niα, ,jβ – зміна числа сусідів певного ти- |
||
пу, Vα ,β |
– відповідні енергії зв’язків. Надлишкові енергії εα ,β |
, а та- |
|
i,j |
|
i, j |
|
|
|
|
221 |
кож енергії Viα,j,β , пов’язані зі значеннями теплоти випаровування,
розраховувалися в [33-35] через табличні і діаграмні дані [36] згідно до формул для регулярних розчинів [37]. Відповідно, в програмах передбачувався підрахунок числа зв’язків усіх типів в попередньому
і можливому новому положенні атома. Крок за часом τmin розраховувався, виходячи з умови, що якщо з випадково обраним атомом одночасно може відбутись декілька подій, то сума ймовірностей їх здійснення не може перевищувати одиницю.
Для апробації моделі граткової рідини у [34] проведено аналіз атомної будови поверхні кристала за результатами моделювання росту однокомпонентного кристала. Початковий розмір кристала з прямою сходинкою складав 80х80х10 міжатомних відстаней, а над ним задавалася досить протяжна область з умовно “ рідких” атомів. У двох напрямках (x, y) застосовувалися періодичні граничні умови.
Приклади отримуваних структур міжфазної границі для перерізу, що містить сходинку, показані на рис. 6.6 (див. розділ 6). При наближенні до точки розупорядковуючого переходу (рис. 6.6, с) сходинка стає майже невиразною. При цьому на поверхні виникає і зникає безліч дірок, а також двовимірних зародків різного розміру, а сама міжфазна межа становить вже область шириною в декілька міжатомних відстаней.
Урахування енергії активації u=2nlηϕ для приєднання атомів до
кристала (даним рис. 6.6 відповідає значення u/2ϕ=0.5) призводить при розрахунках до структур зі значно меншою шорсткістю граней, а температура переходу від гладенької до шорсткої в атомному масштабі міжфазної межі істотно підвищується. При величині відношення
u/2ϕ=1 розупорядковуючому поверхневому переходу відповідає значення енергії зв'язку ϕ/kT 0.34, тобто значення параметра Джек-
сона для переходу від гранних до округлих форм росту α 1.36, що відповідає експериментальним даним для бінарних систем [38]. Автори [34] знайшли, що для гладеньких сходинок швидкість росту є нелінійною функцією від пересичення. При значній шорсткості гра-
ней Vst=βstσ, де βst – кінетичний коефіцієнт для сходинки, σ=Δμ/kT.
Величина βstτ0/a 10-3, тобто βst 1 м/с, що близько до відомих експериментальних значень кінетичного коефіцієнта росту кристалів за
нормальним механізмом.
222
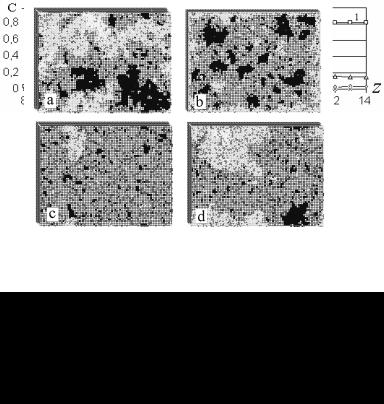
Результати моделювання росту кристалів зі змішаних розплавів за моделлю граткової рідини отримані в [34]. В якості об'єктів обрані сплави систем, у яких форми та кінетика росту первинних кристалів вісмуту [38] по-різному змінюються при зміні складу розплавів і рівноважної температури (температури ліквідусу). Для розрахунку енергій взаємодії атомів використовувалися табличні та діаграмні дані.
Рис. 7.8. Зображення міжфазної поверхні,
a, b, c – ріст з розплавів Bi-Sn, вміст Sn – 5, 10 і 15 ат. % відповідно;
d – Bi+15 ат.% In (у верхньому шарі є надкритичний двовимірний зародок).
Рис.7.9. Розподіли концентрацій компонентів і частки кристалічної фази в послідовних атомних площинах:
а, b – для системи Bi-Sn (10 і 15 ат. % Sn відповідно), c – Bi+15 ат. % In; 1 – частка рідкого А-компонента (Bi), 2 – В-компонента,
3 – вакансій, 4 – частка кристалічної фази (Bi).
Рис. 7.8 зображує структуру поверхні кристалів, отриману в результаті моделювання [34] росту первинних кристалів з розплавів систем Bi-Sn, Bi-In. А на рис. 7.9 наведені результати пошарового аналізу розподілу компонентів, кристалічної фази і вакансій. З рис. 7.8 c, d можна бачити, що у разі системи Bi-Sn (теплота випа-
223

ровування олова більша, ніж у індію) кристалічні кластери в поверхневому шарі крупніші та мають правильнішу форму. А з рис. 7.9 можна бачити, що ширина міжфазної межі істотно зменшується при збільшені концентрації другого компонента. Дані моделювання в цілому узгоджуються з експериментальними даними щодо відмінностей у формах і кінетиці росту первинних кристалів вісмуту у цих системах [38].
7.2.5. Кінетичне Монте-Карло моделювання спрямованого твердіння
В [15] наведено результати вивчення розвитку хвильових збурень форми фронту кристалізації при спрямованому твердінні шляхом кінетичного Монте-Карло моделювання. На рис. 7.10 показано, як при спрямованому твердінні атоми домішки (темно-сірі) скупчуються перед поверхнею зростаючого кристала. А на рис. 7.11 наведені перерізи змодельованих зразків поблизу фронту кристалізації.
Рис. 7.10. Фрагмент перерізу змодельованого зразка.
Розподіл концентрації, що відповідає рис. 7.10, автори [15] порівняли з аналітичним розв’язком одновимірної дифузійної задачі спрямованого твердіння (формула (6.39)) і визначили коефіцієнт ди-
фузії для моделі (D@ 1.2×10–5 см2/с). Це дозволило їм провести порівняння розвитку комірчастих структур з результатами теорії малих збурень [39] і дійти висновку, що збурення з досить великими у атомному масштабі довжинами хвиль розвиваються у відповідності до теоретичних передбачень.
Моделювання виявило існування нижньої межі довжин хвиль, для яких збурення поверхні можуть розвиватись з утворенням стійкої комірчастої структури: ~30 міжатомних відстаней; збурення меншого розміру руйнуються флуктуаціями росту. Передбачена теорією [39] стійкість фронту при дуже великих швидкостях протягування зразка не підтверджується моделюванням. Локальні флуктуації швидкості росту приводять до випадкових збурень поверхні, а структура кристала, який утворюється за таких умов, є украй недосконалою.
224
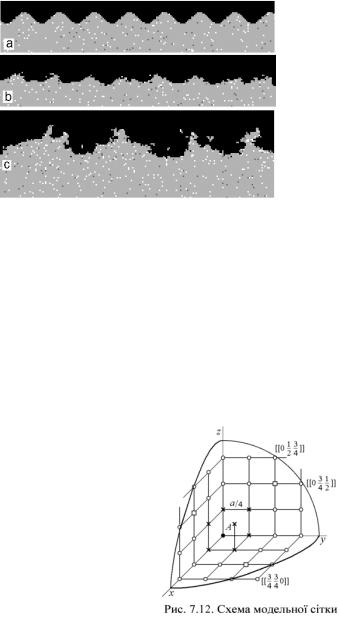
Рис. 7.11. Еволюція збурень міжфазної межі,
а – початкові збурення з довжиною хвилі l=25 міжатомних відстаней (а), b – їх еволюція; с – зростаючі збурення (l= 50 міжатомних відстаней, швидкість росту V=5.4×106 міжатомних відстаней за 1с).
7.2.6.Кінетичне моделювання процесу осадження плівок
згазової фази
Вроботі [40] для кінетичного моделювання конденсації плівок була використана модель, в якій немає умовного розділення атомів на “ тверді” чи “ рідкі”, а вакансії утворюються внаслідок теплового руху атомів за рахунок їхньої дифузії з поверхні. Моделювання проводиться на модельній сітці (рис. 7.12) з кроком за усіма напрямками меншим ніж міжатомна відстань (у 4 рази менше за очікуваний параметр кристалічної гратки a). На рис.
7.12показані лише вузли, що
належать площинам xy, yz, zx. Атомні місця, що відповідають ГЦК гратці, позначені квадратами ( ). Якщо атом А, що розглядається, знаходиться у вузлі [[000]], то сусідній атом може знаходитись на відстані, не ближчій за a/2. Вузли, позначені хрестиками (
). Якщо атом А, що розглядається, знаходиться у вузлі [[000]], то сусідній атом може знаходитись на відстані, не ближчій за a/2. Вузли, позначені хрестиками ( ), заборонені для стрибків у них сусідніх атомів. У якості об’єктів були вибрані речовини Pb, Ni, для
), заборонені для стрибків у них сусідніх атомів. У якості об’єктів були вибрані речовини Pb, Ni, для
яких встановлені експериментальні закономірності щодо зміни механізму конденсації [41].
225

Імовірність атома сісти на підкладку, приєднатись до краплі рідини або кристалика визначалась заданим потоком атомів I до поверхні (0.1÷250 атомів за 1 мікросекунду на одне атомне місце):
pконд.=Iτmin |
(7.29) |
Приєднання допускалось, якщо до відповідного вузла сітки був вільний доступ зверху і жоден з 26 найближчих (заборонених) вузлів сітки не був зайнятим іншим атомом.
Імовірності випарування (зникнення) атомів залежать від їхнього оточення (кількості сусідніх атомів на різних відстанях), а у разі стрибка ще й від його нового оточення. На кожному кроці за часом проводилися тестування усіх атомів щодо стрибків чи випаровування, а також усіх вільних вузлів біля атомів або біля підкладки (на відстані не менш ніж a/2) на можливість заняття їх атомами з падаючого потоку.
Імовірність стрибка визначалася за формулою:
pстриб= τνoexp(–( Enew– Eold) /kT), |
(7.30) |
де Enew та Eold – енергії взаємодії атома з усіма сусідніми атомами в межах сфери обрізання потенціалу парної взаємодії для можливого
нового та попереднього місцеположення розглядуваного атома; νo – частота власних коливань атомів. Якщо нова енергія була менше ніж попередня, стрибок виконувався. А у разі збільшення енергії стрибок виконувався, якщо генероване випадкове число було менше за цю імовірність. Підрахунок енергій проводився за 176 позиціями можливих положень сусідніх атомів. Оскільки число різних можливих переміщень атома дуже велике, провести нормування ймовірностей складно. Тому крок за часом обирався таким чином, щоб подальше його зменшення не впливало на результати обчислень.
Рис. 7.13. Острівки свинцю на підкладці.
226

На рис. 7.13 наведене зображення початкової стадії конденсації крапель свинцю на підкладку за температури Т=610 К. Доки поверхнею підкладки переміщуються поодинокі атоми, це поверхневий газ. Час очікування зародження крапель залежить від падаючого потоку І. Цей час дуже великий при малій інтенсивності потоку, або при низькому значенні коефіцієнта взаємодії bs з підкладкою. На рис. 7.14 показана зростаюча крапля свинцю (переріз, що містить атоми з двох найближчих площин модельної сітки). Можна бачити, що контактні кути крапель з підкладкою збільшується при збільшенні розміру краплі.
Рис. 7.14. Зростаюча крапля свинцю,
Т=610 К, І =32.9 мкс-1, t = 0, 0.125, 0.161 і 0.204 мкс відповідно
(розмір клітинки дорівнює параметру гратки a).
На рис 7.15 показані краплі свинцю, осаджені за температури T = 580 K. На послідовних зображеннях можна бачити парне зіткнення рідких острівків, що призводить до їхнього злиття. Крапля, яка внаслідок цього утворилась, вочевидь, вже перевищує критичний розмір, – в ній можна бачити кристалічну ГЦК-фазу (невелика кількість атомів, позначена темнішим відтінком).
Рис. 7.15. Злиття крапель свинцю та їх подальша кристалізація,
T = 580 K.
Отримані в [40] значення температури Tg зміни механізму конденсації острівкових плівок (пара– кристал на пара– рідина– кристал)
Tg ≈0.77 TПЛ для Pb і Tg ≈0.88 TПЛ для Ni більше за експериментальні значення (0.69 TПЛ для Pb і 0.66 TПЛ для Ni [41]). Це, скоріш за все, пов’язано із розрахунком рушійної сили кристалізації (різниці хімічних потенціалів двох фаз) за спрощеною формулою, Δμ= Н Т/Тпл, яка не враховує різниці теплоємностей фаз.
227
7.3. Особливості застосування методу молекулярної динаміки у разі фазових перетворень.
7.3.1.Застосування методу молекулярної динаміки
врізних ансамблях
Для моделювання фазового переходу рідина-тверде тіло найчастіше використовують NPT ансамбль (канонічний ансамбль Гіббса), тобто підтримують постійними число частинок, тиск і температуру зразка [6,42]. У дійсності фізичні процеси, спостережувані в реальному експерименті, зазвичай відбуваються за постійної температури й тиску. Для утримання постійних тиску й температури необхідно масштабувати (підстроювати) розміри модельного боксу й швидкостей атомів [43,44].
Інший широко використовуваний термодинамічний NVT ансамбль (канонічний ансамбль Гельмгольца), де замість тиску зберігається об’єм (середня щільність) зразка. Об’єм основної комірки можна задавати в початкових умовах. Визначені залежності тиску від об’єму, Р(V), та енергії від об’єму, U(V), дають рівняння стану; вони дозволяють визначити стисливість системи, коефіцієнт розширення та його температурну залежність. Однак нерідко для молекулярнодинамічного моделювання використовується NVE ансамбль (мікроканонічний ансамбль), де зберігається повна енергія системи. Використання цього ансамблю виявляється достатнім для багатьох "прикладних" досліджень у разі, якщо врахування термодинамічних особливостей не дуже важливе. Цей ансамбль реалізує консервативну класичну механічну систему, тому для його реалізації не потрібні спеціальні засоби, які необхідні для реалізації інших ансамблів. Також застосовують NPE ансамбль (ізобарно-ізоенергетичний ансамбль) й ансамбль з N≠Const (великий канонічний ансамбль), в якому можливо здійснювати обмін частинками з термостатом.
Наведемо декілька найбільш застосовуваних методів для одержання постійними необхідних параметрів.
NVT ансамбль:
1. Метод термостабілізації, полягає в тому, щоб періодично перепризначувати атомам швидкості навмання – відповідно до розподілу Максвелла-Больцмана [45]. Перепризначення може бути зроблено для окремих атомів, або для всієї системи в цілому.
228
2. Алгоритм Берендсена [46]: в цьому методі реалізується помноження швидкостей всіх атомів на певний коефіцієнт λ на кожному кроці за часом, або через кожні декілька (5÷10) кроків:
|
|
τ |
|
T |
|
1/2 |
|
||
λ = 1 |
+ |
|
− 1 |
|
, |
(7.31) |
|||
|
|
||||||||
|
|
tc Tм |
|
|
|
|
|||
де τ – крок за часом; tc – характерний час в одиницях t0 (який потрібно підібрати); Тм – миттєва температура; Т– задана температура.
3. Алгоритм Нозе-Гувера полягає у введені в динамічні рівняння додаткової змінної [47,48], яка пов’язана з тепловим резервуаром:
|
& |
|
|
pi |
|
|
|
|
|
|
|
|
|
|
= m |
, |
|
|
|
|
|
(7.32,а) |
|||||
|
|
ri |
|
|
|
|
|
||||||
|
& |
= fi |
|
− ξ pi , |
|
|
|
|
(7.32,б) |
||||
|
|
pi |
|
|
|
|
|
||||||
|
∑ia pi2a /m − gkBT |
2 |
|
∑ia pi2a /m |
|
2 |
T |
|
|
||||
ξ = |
|
≡ ηT |
|
|
|
|
−1 |
= ηT |
|
−1 |
, (7.32,в) |
||
|
|
gk |
T |
T |
|||||||||
|
Q |
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
B |
|
|
|
|
|
|
де pi (pia) – імпульс і-ї частинки, p&i – похідна за часом від імпульсу, fi
– сила яка діє на і-ту частинку, ξ – коефіцієнт тертя, який може змінюватися з часом; Q – теплова інерція, цей параметр може бути замінений на ηT=gkBT/Q, тобто на параметр релаксації для теплових коливань; g=3N є числом ступенів вільності, Tм – миттєва температура. В цьому випадку функція розподілу для ансамблю пропорційна
exp(–W/kBT), де |
W = H + |
1 |
3N kB Tξ2 /ηT2 , H = ∑ pia2 /2m + U , |
||
|
|||||
|
2 |
|
ia |
|
|
|
|
|
|
|
|
U– потенціальна енергія системи. |
|
|
|||
Енергія системи є залежною від часу |
|
||||
H = |
∑ piapia /m − ∑ fiaria |
= −ξ∑ pia /m . |
(7.33) |
||
& |
& |
|
& |
2 |
|
|
ia |
|
ia |
|
|
Якщо Tм>T, тобто система є занадто гарячою, коефіцієнт тертя ξ стає додатним і система починає охолоджуватись. Якщо система є занадто холодною, відбувається зворотне: коефіцієнт тертя стає від’ємним і система починає нагріватися.
Існують інші алгоритми утримання постійної температури наприклад: алгоритм Андерсена [45], алгоритм Ланжевена [49]. Алгоритми Андерсена та Ланжевена полягають в тому, що система, має
229
постійну взаємодію з фіктивними (віртуальними) частинками і обмінюється з ними енергією. Практично ці взаємодії призначені для заміни швидкості атомів на нові швидкості відповідно до розподілу Больцмана за енергіям [45] й розподілу Гаусса за швидкостями [49].
NРT ансамбль:
Андерсен [45] запропонував метод стабілізації тиску, у якому до системи додається додаткова змінна V – об’єм комірки, що моделюється. Це еквівалентно дії поршня на реальну систему. Поршень має “ масу” Q (остання має розмірність кг×м–4 ) й пов’язану з нею кінетич-
ну енергію EV = 0.5QV |
та потенціальну енергію, пов’язану з об’ємом |
& 2 |
|
розрахункової комірки UV=PV, де P – потрібний тиск. Потенціальна й кінетична енергії атомів пов’язані ще зі змінними r та v, які нормовані згідно з наступними співвідношеннями
r = V1/3s , |
(7.34,а) |
||
v = V |
1/3 |
& |
(7.34,б) |
|
s. |
||
Потенціальна енергія частинок є функцією координат частинок U=U(V1/3s), й відповідно перша складова Лагранжіана (Lv=Ek+EV-U-UV) має наступний вигляд:
2 |
=½mV |
2/3 |
&2 |
(7.35) |
Еk=½m ∑ vi |
|
∑ si . |
ii
Рівняння руху будуть такими:
&& |
|
1/3 |
) - (2 |
& & |
(7.36) |
si = fi /(mV |
|
/3)siV /V , |
|||
&& |
= (PM |
- P)/Q , |
(7.36) |
||
V |
|||||
де &s&i – прискорення і-ої частинки, fі – |
зведена сила, Pм – |
миттєве |
|||
значення тиску. Гамільтоніан цієї системи, H=Ek+EV+U+UV, зберігається, він дорівнює ентальпії з додатковим фактором kBT, пов’язаним з кінетичною енергією флуктуації об’єму. Рівнянням руху задовольняють траєкторії, які відповідають ізобарноізоентальпійному NPH ансамблю.
В роботі [50] описано застосування цього методу в МД моделі. Оскільки диференціальне рівняння для об’єму включено в розширені рівняння руху, сили та тиск розраховуються, використовуючи ненормовані координати. Для розрахунку траєкторії руху атомів автори використовували рівняння для ненормованих швидкостей
230