

Ovru-all
.pdfнентами і ф фазами маємо ф+к×(ф–1) рівнянь, а кількість незалежних величин (концентрацій) запишеться так:
к×ф– ф– к×(ф–1)=к– ф.
Крім того, температура та тиск - також незалежні параметри. Отже, загальна кількість незалежних параметрів, або ступенів вільності системи,
С=к–ф+2.
Ця кількість не може бути від’ємною (С>0). Звідси випливає правило фаз Гіббса
ф≤к+2, |
(2.58) |
яке визначає максимальну кількість фаз, що існують одночасно за умов рівноваги.
Уздовж кривої рівноваги конденсованої однокомпонентної речовини з парою маємо дві фази, а ступінь вільності при цьому один. Довільна зміна тиску повністю визначає необхідну для збереження рівноваги зміну температури (або навпаки). В однокомпонентній системі в рівновазі можуть перебувати і три фази, наприклад: кристал, рідина і газ. Але в цьому стані система не має ступенів вільності. У разі зміни температури або тиску одна із фаз зникає. Це так звана потрійна точка.
2.5.5. Діаграми стану
Діаграмою стану називають сукупність кривих рівноваги фаз.
Діаграми стану розглядаються як у загальному курсі фізики, так і в спецкурсах з термодинаміки та кінетики фазових перетворень. Розглянемо тільки дві діаграми, що відображують рівновагу кристалічних та рідкої фаз.
Діаграма стану двокомпонентної системи типу сигари наведена на рис.2.6. Їй відповідає утворення у твердому стані безперервного ряду твердих розчинів (у рідкому стані теж утворюються розчини за будьяких концентрацій компонентів). Такою, наприклад, є діаграма стану системи мідь-нікель. На шкалах температур цієї діаграми відмічені температури плавлення чистих компонентів. На осі абсцис відкладена концентрація B-компонента. Крива 1 називається лінією ліквідусу,
а крива 2 - лінією солідусу. За температури T1 в рівновазі знаходяться рідина з концентрацією Cр1 і кристал з концентрацією Cкр1. Під час поступового зниження температури кількість рідини в системі змен-
61
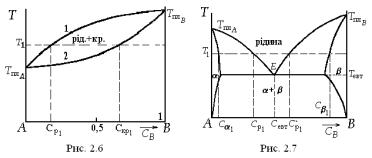
шується, а кількість кристалічної фази зростає, їх рівноважні концентрації змінюються згідно з кривими рівноваги 1 та 2.
Діаграму стану, зображену на рис.2.7, називають діаграмою евтектичного типу. Такою є, наприклад, діаграма стану системи свинецьолово. Тут теж можуть існувати тверді розчини, але лише в тому разі, якщо добавка другого компонента до основного порівняно мала. Якщо концентрація B-компонента в системі, менша від Cевт, за тем-
ператури T1 в рівновазі можуть знаходитися твердий розчин α з концентрацією Cα1 та рідина з концентрацією Cp1 . Якщо ж концентра-
ція перевищує Cевт , у рівновазі будуть перебувати твердий розчин β з концентрацією Cβ1 і рідина з концентрацією Cp` 1 . Точка E зветься
евтектичною. У ній в рівновазі знаходяться рідина і два твердих розчини α і β. Якщо врахувати ще газ з компонентів A і B, який є над розплавом, то стає зрозуміло, що точка E є четверною точкою. Нижче температури Tевт в рівновазі можуть існувати лише тверді розчини та газ. Під час кристалізації сплаву евтектичного складу з розплаву ростуть одночасно кристали α- і β-фаз. Найчастіше вони утворюють регулярні структури − так звані евтектичні колонії.
2.6. Вплив поверхневого натягу на кристалізацію рідин
Поверхневий натяг відіграє дуже значну роль у фазових перетвореннях. Він, наприклад, обумовлює можливість існування метастабільних станів речовини, таких як пересичена пара та перегріта рідина.
Припустимо, що рідина з капіляром, стінки якого вона не змочує, знаходяться в закритій посудині, так що пара над рідиною є рівноважною. Але тиск пари залежить від висоти. Над опуклою поверхнею
62

рідини в капілярі (рівень нижчий, ніж у посудині) він більший за тиск пари над плоскою поверхнею Рo :
Рr=Рo+ρпgh=Рo+ |
ρп |
2σ |
, |
(2.59) |
|
||||
|
ρр |
R |
|
|
де R – радіус кривизни поверхні рідини, ρр – густина розчину.
Це дуже важлива формула. Якщо в посудину, де є насичена пара, внести краплю рідини, вона буде випаровуватись, тому що тиск насиченої пари для краплі більший, ніж тиск насиченої пари для плоскої поверхні (для краплі пара недосичена). Для того щоб у парі виникли краплі рідини, треба досягти такого тиску пари, який буде рівноважним для так званих центрів конденсації, тобто для малих краплинок рідини, які утворюються на частинках пилу. А якщо пил відсутній, то для виникнення рідини необхідне дуже велике пересичення пари.
Формулу (2.59) можна переписати у вигляді
r* = 2σ ρп ,
P − Po ρр
і тоді вона дозволить обчислювати розмір краплі r*, для якої справджується динамічна рівновага між числом молекул, що конденсуються, і числом молекул, які випаровуються за той же самий час. Але ця рівновага не є стійкою. Якщо розмір краплі стане дещо меншим внаслідок флуктуацій кількостей молекул, що конденсуються або випаровуються, то пара над краплею стане ненасиченою: Р=Рr*<Рr’ , і крапля буде далі випаровуватись. І навпаки, якщо розмір краплі збільшиться, вона стане центром конденсації (Р=Рr*>Рr’ ) і її об’єм продовжуватиме збільшуватись.
Поверхневий натяг впливає не тільки на рівновагу рідини та її пари. Для будь-якого фазового переходу першого роду виникнення зародків нової фази пов’язане з утворенням поверхні розділу. Якщо в рідині виникне зародок кристала у формі кулі радіусом r, то вільна енергія Гіббса системи зміниться на величину
G= Nкμк+ Nрμр+4πr2σ
або |
G=–(4πr 3/3Ω)(μр–μк)+4πr2σ, |
|
|
(2.60) |
|
де μк і μр – |
хімічні потенціали твердої та рідкої фази; |
Nк=– Nр; |
(4πr3/3Ω) – |
кількість молекул у зародку; Ω – об’єм однієї молекули; |
|
|
|
63 |
σ – питома вільна поверхнева енергія; 4πr2 – площа поверхні зародка. Перший доданок у формулі (2.60) від’ємний, оскільки для переохолодженої рідини μр>μк. фази; NA – число Авогадро; Т=Тпл– T – переохолодження.
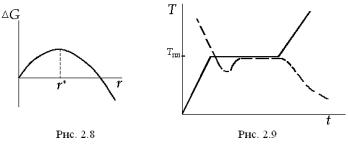
Другий доданок у формулі (2.60) завжди додатний, у випадку малих розмірів зародка він більший за перший доданок. Зміна вільної
енергії Гіббса ( G) системи після виникнення зародка залежить від його розміру. Цю залежність якісно відображує рис. 2.8. Якщо розміри зародків малі, величина G додатна, і вона тим більша, чим більший розмір r. Існує деякий розмір, будемо називати його критичним розміром і позначати r*, при досягненні якого величина G сягає максимуму, а потім починає зменшуватись. Якщо розмір зародка перевищить величину r*, то приєднання до нього нових молекул буде призводити до зниження вільної енергії системи. Такий зародок називають уже центром кристалізації. Критичний розмір r* можна знайти з умови екстремуму функції G(r). Виконаємо диференціювання по r і прирівняємо першу похідну до нуля. Тоді
r*= |
2Ωσ |
|
= |
2VμσTпл |
. |
(2.61) |
μр − μ |
|
|
||||
|
к |
|
L T |
|
||
|
|
|
|
|
|
|
Хімічний потенціал однокомпонентної системи – це вільна енергія Гіббса в розрахунку на одну молекулу, в умовах рівноваги (температура плавлення) хімічні потенціали двох фаз рівні. Для переохолодженої системи
Gрμ–Gкμ=Hр–TSр–(Hк–TSк)= Hμ T Sμ,
де Н – ентальпія; S – ентропія; Н=L – теплота фазового переходу (теплота кристалізації) в розрахунку на один моль речовини. З умови
64

рівності вільних енергій Gрμ=Gкμ за температури плавлення Тпл випливає зв’язок: Нμ=Тпл Sμ. Тому
μр–μк=(Gрμ–Gкμ)/NA= L T = ΩL T ,
NATпл VμТпл
де L – теплота кристалізації; Vμ – молярний об’єм кристалічної фази. Вираз (2.61) називають формулою Томсона. Вона дозволяє визначати розмір кристала, який знаходиться в рівновазі (нестійкій рівновазі) з переохолодженою рідиною. Підставивши вираз для r* у формулу (2.60), знайдемо роботу утворення зародка критичного розміру:
G(r*)=– (8/3)πσ(r*)2+4πr*σ=(4/3)πσ(r*)2. |
(2.62) |
У випадку малих переохолоджень рівноважний розмір кристалів великий і робота утворення таких великих центрів кристалізації дуже значна, а ймовірність їх виникнення за скінченний час надто мала. Досліди з вивчення так званої гомогенної кристалізації, тобто кристалізації малих крапель, вільних від забруднень та завислих у рідині малих твердих частинок, показали, що для утворення центрів кристалізації потрібні дуже великі переохолодження, які становлять
значну частину від температури плавлення, Т=Тпл-Т≈ 0,2Тпл. Імовірність утворення центрів кристалізації пов’язана ще й з
об’ємом рідини. Під час швидкого охолодження малих крапель різних речовин в багатьох із них кристалічні центри зовсім не утворюються, а рідина переходить в аморфне тверде тіло. Аморфні матеріали, отримані методом швидкої кристалізації, нині широко застосовуються в різних галузях науки і техніки. Метод охолодження рідини з надвисокими швидкостями для отримання металів у метастабільних станах (нерівноважних для даних умов) одними з перших застосували вчені Дніпропетровського університету – І. В. Саллі, М. Й. Варич, І. С. Мірошниченко.
Під час охолодження досить великих об’ємів рідини значних переохолоджень досягти не вдається, тому що центри кристалізації виникають не в об’ємі рідини, а на стінках посудини або на твердих частинках, що є в рідині. У цьому разі говорять про гетерогенну кристалізацію. За звичайних умов майже завжди має місце гетерогенна кристалізація, тому що робота утворення зародків на стінках набагато менша, ніж робота утворення зародків в об’ємі речовини. Фільтруванням рідини можна впливати на кількість твердих домішок і на величину переохолодження, необхідного для початку кристалізації.
65
До збільшення величини переохолодження призводить також перегрів рідини вище температури плавлення перед початком охолодження.
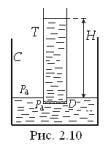
Без переохолодження рідини кристалізація не розпочинається, тому криві плавлення і твердіння (залежності температури від часу для нагрівання й охолодження) речовини якісно відрізняються (рис. 2.9). Крива плавлення (вона показана на рисунку суцільною лінією) має горизонтальну ділянку, яка відповідає значенню Т=Тпл, що пов’язано з поглинанням теплоти кристалізації (теплота, яка вводиться в систему, іде на збільшення внутрішньої потенціальної енергії). Пунктирна крива охолодження свідчить про те, що кристалізація розпочинається тоді, коли температура речовини менша, ніж температура плавлення. За рахунок теплоти кристалізації речовина розігрівається майже до температури плавлення. Коли кристалізація закінчується, температура починає спадати.
На процес кристалізації істотно впливає кількість центрів кристалізації. Якщо їх утворюється багато, то у твердому стані речовина має полікристалічну структуру. Під час кристалізації з одного центра, що практично важко здійснити, речовина стає монокристалом. Монокристали використовуються для виготовлення більшості електронних приладів. Це стимулює розвиток фізики росту кристалів та промисловості вирощування монокристалів.
Вивченню кристалізації рідин значно сприяли роботи академіка В. І. Данилова та його учнів, які були розпочаті до Другої світової війни в м. Дніпропетровську (в університеті та фізико-технічному інституті), а потім продовжені в Інституті металофізики ім. Г.В. Курдюмова НАН України (м. Київ, професори Д.Ю. Овсієнко, А.Г. Леснік, Д.С. Камінецька) та багатьох інших наукових центрах.
2.7.Явища, пов’язані з утворенням розчинів
2.7.1.Тепловий ефект утворення розчину
Розчином називають однорідну суміш двох або більшої кількості речовин. Якщо однієї з цих речовин значно більше, ніж інших, то вона зветься розчинником, а інші – розчиненими в розчині. Розчини, які складаються з двох компонентів, називаються бінарними. Залежно від свого фазового стану розчин може бути рідким або твердим.
Рівноважний стан речовини за постійної температури та об’єму визначається мінімумом вільної енергії Гельмгольца F=U−TS. Вільна енергія зменшується, якщо зменшується внутрішня енергія або збільшується ентропія. Під час зміщування атомів різних речовин ент-
66
ропія завжди збільшується, тому що збільшується термодинамічна ймовірність системи (згідно із формулою Больцмана: S=klnWT). Тому розчин може утворитися і в тому разі, коли внутрішня енергія збільшується. Залежно від знака зміни внутрішньої енергії у процесі виникнення розчинів теплота може виділятися (екзотермічний процес) або поглинатися (ендотермічний процес).
Зміна внутрішньої енергії в разі утворення бінарного розчину пов’язана з числом різнойменних пар атомів NАB (А і В – назви компонентів) та надлишковою енергією їх взаємодії, яка визначається формулою
εAB=VAB−(VAA+ VBB)/2, |
(2.63) |
де VAA, VBB i VAB – енергії взаємодії відповідних пар атомів (вони всі від’ємні, тому що за нуль приймається потенціальна енергія вільних атомів). Якщо надлишкова енергія εAB дорівнює нулю, то розчин на-
зивається ідеальним. У процесі його утворення теплові ефекти від-
сутні. Якщо 2 VАВ > VАA + VBB, тo теплота змішування W=−NAB εAB більша за нуль (екзотермічна реакція утворення розчи-
ну, Q<0). Якщо ж 2 VАВ < VАA + VBB , то утворення розчину веде до збільшення внутрішньої енергії (W= – NAB εAB<0 − теплота погли-
нається, Q>0 , а реакція утворення розчину ендотермічна). Такі розчини утворюються тільки за досить високих температур.
Неідеальні розчини, як правило, дещо впорядковані. Якщо εAB<0, енергетично вигідно, щоб атоми одного компонента мали у якості найближчих сусідів атоми іншого компонента, при цьому число NAB
збільшується. І навпаки, якщо εAB>0, то вигідніше, щоб кожний атом мав найближче оточення з таких же атомів, як і він сам. Але при цьому зменшується ентропія. Рівноважному станові відповідає деякий визначений рівень упорядкування щодо розміщення атомів.
2.7.2. Осмотичний тиск
Нехай у деяку посудину С (рис. 2.10), в яку налито чистий рідкий компонент А (розчинник), опущена трубка Т, закрита знизу напівпроникною перегородкою D, яка пропускає молекули розчинника і не пропускає молекул компонента В, налитого в трубку T розчину. Тиск на стінки трубки Т чинить і розчинник, і розчинена речовина. Сума цих тисків також дорівнює сумі атмосферного й гідростатич-
67
ного тисків: Pa+ρgh (ρ – густина розчину). Тиск розчинника над перегородкою D
PA=Pa+ρgH−П, |
(2.64) |
де Н – висота рідини в трубці; а П – так званий осмотичний тиск розчиненої речовини. Якщо тиск PA А-компонента менший ніж Pa, якому дорівнює тиск чистого розчинника знизу від перегородки, то молекули розчинника будуть переходити крізь перегородку з посудини у трубку Т. Цей процес буде тривати доти, доки тиск PA не досягне величини Pa. З цієї умови випливає, що динамічній рівновазі на напівпроникній перегородці D відповідає визначена висота
стовпчика розчину в трубці Т:
H=П/ρg. |
(2.65) |
У випадку досить малої концентрації у розчині компонент В можна розглядати як ідеальний газ (тобто нехтувати взаємодією молекул). Тому складову тиску розчиненої речовини можна розрахувати за газовим законом (формула Вант-Гоффа)
П= PB=(mB/μB)RT/V=(ρB/μB)RT, |
(2.66) |
де mB та μB – маса та молярна маса компонента В відповідно; ρB – його концентрація. У випадку концентрації В-компонента в декілька відсотків (наприклад, розчин солі або цукру у воді) цей тиск досягає десятків атмосфер. Для внесення ясності щодо сенсу явища осмосу підкреслимо, що розчинник у розчині може знаходитись під значним від’ємним тиском. Атмосферному тискові відповідає певна густина розчинника. Густина розчинника у розчині менша ніж це значення. Отже, розчинник знаходиться в розтягнутому стані.
2.7.3. Закони Рауля і Генрі
Перший закон Рауля і закон Генрі
Зрозуміло, що внаслідок внесення домішки деякої речовини в розчинник динамічна рівновага між ним та його парою порушиться. Кількість молекул, що вилітають з розчинника, стане меншою, і цій кількості відповідатиме менший рівноважний тиск пари. Для його знаходження уявимо собі, що посудина С з трубкою Т (рис. 2.10) знаходяться всередині великої закритої посудини. Тиск пари в такій
68
системі (ми розглядаємо її рівноважний стан) залежить від висоти. На висоті Н тиск
Р=Ро–ρпgH, |
(2.67) |
де Ро – тиск пари над чистим розчинником, а ρп – густина його пари. Підставимо в останню рівність вираз висоти H через осмотичний тиск (2.65) і врахуємо співвідношення (2.66). Тоді
Po − P |
= ρп g |
П |
= |
ρпρBRT μ A = |
ρB μ A = |
υB |
, |
||||||
P |
|
|
|||||||||||
P ρg |
ρ |
п |
RTρ μ |
B |
μ |
B |
ρ υ |
A |
|
||||
o |
o |
|
|
|
|
|
|
||||||
де Ро =ρпRT/μA; νA i νB – число молів компонентів А і В відповідно.
Для малих концентрацій В-компонента νA≈νA+νB, тому останню рівність можна переписати у вигляді
Po − P |
= |
|
υB |
|
. |
(2.68) |
|
P |
υ |
A |
+ υ |
|
|||
o |
|
|
|
B |
|
||
Це формула першого закону Рауля: відносне зниження тиску пари розчинника над розчином прямо пропорційне відносній молярній
концентрації розчиненої речовини.
Генрі запропонував аналогічний зв’язок для концентрації розчиненої речовини і її тиску над розчином:
υB |
~ PB . |
(2.69) |
|
υA + υB |
|||
|
|
Прочитаний справа наліво закон Генрі, тобто
РВ= Po |
υB |
, |
(2.70) |
|
|||
B |
υA + υB |
|
|
називають законом Рауля для розчиненої речовини. Стосовно ідеальних розчинів
закони Рауля і Генрі справедливі для всіх концентрацій компонентів,
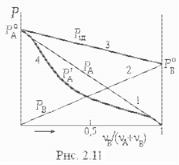
а у випадку реальних розчинів − тільки для малих концентрацій одного із двох компонентів. На рис. 2.11 показана залежність парціальних тисків компонентів від їх концентрації в розчині. Прямі 1 і 2 відповідають випадку ідеального розчину. Пряма 3, яка з’єднує точки
PAo і PBo (тиски пари над чистими компонентами), показує сумарний тиск над ідеальним розчином. Крива 4 відображає залежність від концентрації тиску пари розчинника PA' у випадку реального розчину.
69
Другий закон Рауля
Другий закон Рауля встановлює залежність температури кипіння від концентрації розчиненої речовини в розчині.
Кипіння починається тоді, коли тиск пари над рідиною досягає значення зовнішнього (атмосферного) тиску. Після внесення другого компонента в рідину тиск пари розчинника знижується. Тому кипіння стає можливим лише за більш високої температури, яка забезпечить підвищення тиску пари розчинника до значення зовнішнього тиску.
З рівняння Клапейрона-Клаузіуса (2.46) та першого закону Рауля випливає, що
Tкип= |
RT 2 |
|
υB |
= E |
mB |
/μB |
, |
(2.71) |
λμ |
|
υA + υB |
mA |
/μ A |
де літерою Е позначена так звана ембуліоскопічна стала, E=RT2/λμ, λμ – теплота випаровування в розрахунку на один моль речовини,
υB=mB/μB, υA=mA/μA.
Третій закон Рауля
Третій закон Рауля, який встановлює зв’язок між зниженням температури плавлення та концентрацією розчиненої речовини в рідкому розчині, має такий вигляд:
Tпл=K |
|
υB |
|
, |
(2.72) |
|
υ |
A |
+ υ |
|
|||
|
|
|
B |
|
||
де К= RT2/L – кріоскопічна стала; L – теплота плавлення, віднесена до одного моля. Він випливає із першого та другого законів Рауля.
2.7.4. Парціальні термодинамічні функції
Парціальні функції зручні для теоретичного опису розчинів. Докладно це питання описано в [1]. Розглянемо найперше залежність об’єму розчину від його складу (рис. 2.12). Вона дана кривою V(С). Для ідеального розчину об’єм залежить лінійно від концентрації між значеннями об’ємів для чистих компонентів VA0 та VB0. Парціальні об’єми визначаються відрізками, що відсікаються на осях ординат
(VA та `VB) лінією, дотичною до кривої V(С) в точці, що відповідає розглядуваній концентрації (відносна молярна концентрація хВ=СВ=υА/(υА+υВ), υА та υВ – кількості молей компонентів).
70