

Ovru-all
.pdfДля дослідження металів розроблено багато методів розрахунку потенціалів міжатомної взаємодії, які базуються на законах квантової механіки.
Потенціали парної взаємодії, розраховані за методом псевдопотенціалу.
Відомо, що об’єм металу 1см3 складається з ~1023 іонів й вільних електронів. Для атомів, електронні оболонки яких містять велику кількість електронів, точний розв’язок рівняння Шреденгера отримати неможливо (точний розв’язок існує для атомів, у склад яких входять всього декілька електронів). Одним з методів розрахунку потенціалів міжатомної взаємодії, який базується на законах квантової теорії, є метод псевдопотенціалу. Теорія псевдопотенціалу враховує три фундаментальні фізичні наближення. Перше – це наближення самоузгодженого поля, в якому взаємодія між електронами описується деяким усередненим потенціалом, який залежить від стану електронів. Друге наближення полягає в розділенні всіх електронних станів на внутрішні й зовнішні. Третє наближення полягає у використанні теорії малих збурень для знаходження хвильових функцій і відповідних електронних станів.
Рівняння Шредингера розглядається у наближенні самоузгодженого поля
Hyi = [T +U(R)]yi = Eiyi , |
(7.53) |
де Н – гамільтоніан системи, U(R) – самоузгоджений потенціал, діючий на вільний електрон, Т= –(ћ2/2m)Ñ2 – оператор кінетичної енергії, Еі – енергія і– го стану, yі – хвильова функція і– го електрону. Вимога ортогональності хвильових функцій вільних електронів до хвильових функцій внутрішніх оболонок призводить до появи поправки до потенціалу U(R), новий ефективний потенціал зветься псевдопотенціалом. Як правило, поправку вводять напівемпіричним шляхом, будують багато версій модельного потенціалу і обирають з них такий, що якнайкраще відповідає характеристикам даного металу. Для розрахунків застосовувались наступні модельні псевдопотенціали:
1.Псевдопотенціал порожнього іонного остову Ашкрофта [64], в подальшому вдосконалений щодо лужних металів Сінгві, Шью,
Тосі [65].
2.Модельний нелокальний псевдопотенціал Хейне-Абаренкова- Анімалу, який за виглядом більш наближений до псевдопотенціалу розрахованому з перших принципів [66].
241
3.Двопараметричний модельний псевдопотенціал Краско-Гурского
[67].
4.Модельний псевдопотенціал Лерібо-Анжеля, зручний для розрахунків властивостей легких металів з сильною обмінною взаємо-
дією [68].
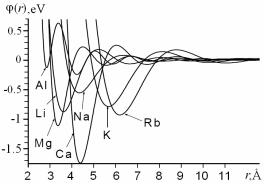
На рис. 7.20 наведено ефективні парні потенціали міжатомної взаємодії для металів Al, Li, Mg, Ca, Na, K, Rb, розраховані за методом псевдопотенціалу [69].
Рис. 7.20. Ефективні парні потенціали деяких чистих металів в кристалічному стані [69].
На форму ефективного потенціалу взаємодії атомів суттєво впливає як вибір псевдопотенціалу, так і метод врахування обміннокореляційних поправок. Тому виникає проблема вибору потенціалу оптимальної версії для вивчення фізичних характеристик певних систем. Рис. 7.21,а демонструє наскільки відрізняються ефективні потенціали взаємодії атомів, для розрахунку яких використанні різні обмінно-кореляційних поправки.
Розрахунки міжатомної взаємодії в рамках теорії функціоналу густини (ТФГ).
Як зазначалось вище, в методі псевдопотенціалу є проблеми з розв’язком рівняння Шреденгера, особливо у випадку атомів, що складаються з великої кількості електронів. В роботах Кона і Хоенберга й Кона і Шема [70,71] було запропоновано для розрахунку основних характеристик системи використовувати електронну густину. Сучасні методи першопринципних розрахунків потенціалів взаємодії атомів виходять із теорії функціоналу електронної густини. Опис
242
сутності методу теорії функціоналу густини (ТФГ) є в роботах [42, 72-74]. Для металів в [75] запропонована наступна схема пошуку електронної густини. В початковому стані моделі задається набір пробних хвильових функцій електронів й пробна початкова електро-
нна густина ρ0(r), яка вводиться у Гамільтоніан. Потім хвильові функції уточнюються методом ітераційної діагоналізації матриці, й розраховується нова електронна густина ρ(r). Початкова густина ρ0(r) й
нова ρ(r) змішуються, і розраховується новий ефективний потенціал. Процедура змішування залежить від того, яка задача розв’язується: моделювання фізичних процесів в об’ємі, на поверхні зразку або дослідження окремих молекул. Ця ітерація повторюється, доки зміни
електронної густини стануть незначними (ρn+1(r) ρn(r)).
Метод “ab-initio” передбачає розрахунки електронної густини на кожному часовому кроці молекулярної динаміки (див. у [42]). У роботі [75] такі розрахунки проведені для рідких Li, Na, Al, Cu, V, Ge, при невеликій кількості атомів в основній розрахунковій комірці < 100. Метою розрахунків було отримання рівноважної структури. Розраховані парні функції радіального розподілу атомів задовільно узгоджуються з експериментальними даними.
Методи зануреного атома
Перший варіант методу зануреного атому (МЗА) було розроблено в роботі [76]. Він призначався для розрахунку потенціалів взаємодії атомів у речовинах з металевим типом зв’язку. Метал, як відомо, являє собою сукупність позитивних іонів, які знаходяться в середовищі вільних електронів. Екранування іонів вільними електронами впливає на кулонівське відштовхування між ними.
Повна енергія системи атомів U в МЗА складається з суми парних взаємодій атомів й з суми енергій занурення атомів в електронне середовище
U = |
1 |
∑ ϕ |
|
(r ) + ∑ F (ρ |
), |
(7.54) |
|
|
ij |
||||||
|
2 i,j |
ij |
i i |
|
|
||
|
|
|
i |
|
|
||
де U – повна енергія взаємодії атомів в системи, ϕij(rij) – потенціал взаємодії між атомами i та j, Fi (ρi ) – енергія, пов’язана із зануренням атому і в електронне середовище в точці rі з електронною густиною ρi. Електронна густина знаходиться як лінійна сума локальних внесків електронної густини від усіх сусідніх атомів j
243
ρi = ∑ f j (rij ) , |
(7.55) |
j¹i |
|
де f j (rij ) – електронна густина, яка створюється атомом j в точці з
координатами атома i, як функція відстаней rij між ними; ця функція є короткодіючою й монотонно спадаючою, вона локалізована в області декількох координаційних сфер. В МЗА вважається, що розподіл електронної густини сферично усереднений. Зрозуміло, що першу суму у (7.54) розраховують у межах певної сфери обрізання потенціалу міжатомної взаємодії. А результати розрахунків представляють як залежність енергії системи (у розрахунку на один атом) від параметру кристалічної гратки, що еквівалентно рівнянню стану.
Модифікований метод зануреного атому (ММЗА) є напівемпі-
ричним методом з єдиним формалізмом для різних кристалічних структур (ГЦК, ОЦК, ГПУ, структури алмаза і навіть кристалів інертних газів). Баскес [77-81] першим запропонував MМЗА, та наступне його удосконалення. ММЗА являє собою також розвиненням МЗА на випадок кутової залежності вкладу в електронну густину від різних атомів. Кутова залежність електронної густини пов’язана з відхиленням поверхні Фермі в металах в кристалічному стані від сферичної форми, що є причиною появи нецентральних сил.
Повна енергія Е системи атомів в MМЗА також як і у МЗА апроксимується як сума енергій атомів. Енергії атомів і складаються із суми енергій парної взаємодії ϕij(rij) з атомами j, що оточують розглядуваний атом (rij – відстані між атомами i та j), й енергії занурення в електронне середовище
E = |
1 |
∑ϕij (rij ) + Fi (ρi ) , |
(7.56) |
|
|||
|
2 i¹ j |
|
|
де F – функція занурення, ρi – електронна густина у місцеположенні атома і. Енергія занурення Fi ( ρi ) являє собою енергію, необхідну
для вилучення атому і з його місця ri з локальною електронною густиною ρi . Вона розраховується за формулою
Fi (ρi ) = AiE0 ρi ln(ρi ) , |
(7.57) |
де E0 – енергія сублімації, параметр Ai залежить від типу атома і, а електронна густина ρi шукається за досить громіздкими формулами
244
(дивись [81]), які враховують різні функції екранування для декількох координаційних сфер й анізотропні складові функцій електро-
нної густини. Парний потенціал ϕij(rij) залежить від міжатомної відстані, від природи взаємодіючих елементів і ще від функції пов’язаної з електронною густиною [81].
В деяких роботах, наприклад [82,83], для пошуку сумарної локальної електронної густини з урахуванням кутової залежності вкладу в електронну густину від різних атомів автори користуються трьохчастинковими функціями, такими самими, які використовували Стілінжер та Терзофф для своїх потенціалів. Нижче наведено загальний вигляд функції, подібної до використаної Терзоффом,
|
2 |
)1/2 = (∑[f j (rij )]2 + 2∑∑ f j (rij ) fk (rik ))1/2 . (7.58) |
|
ρi = ( ∑ f j (rij ) |
|||
|
|
j¹i |
k> j j>i |
j¹i |
|
||
Електронна щільність ρi в точці і розраховується як лінійна сума локальних внесків електронної щільності від усіх сусідніх атомів j, та ще враховується взаємне розташування атомів j і k, які входять у сферу розрахунку.
Автори в [81] звірили достовірність розроблених потенціалів чистих Al та Mg і для системи Al-Mg з фізичними властивостями цих речовин та з результатами розрахунків у рамках ТФГ. Методом ММЗА розраховано потенціали міжатомної взаємодії для таких елементів Cu, Ag, Au, Ni, Pd, Pt, Al, Pb і для декількох бінарних систем
[84].
Потенціали для чистого алюмінію
Алюміній є важливим металом. Об'єм виробництва чистого алюмінію або матеріалів на його основі набагато випереджає випуск решти всіх кольорових металів і поступається тільки виробництву сталі. Це обумовлено його фізико-хімічними властивостями, завдяки яким він знайшов широке застосування в авіа- й машинобудуванні, електротехніці і т. ін. На рис. 7.21 наведені декілька ефективних потенціалів міжатомної взаємодії для чистого алюмінію, розрахованих за методом псевдопотенціалу й за МЗА, які запропоновані різними авторами у різні роки.
На рис. 7.21,а наведені ефективні потенціали міжатомної взаємодії, розраховані в рамках теорії псевдопотенціалу (псевдопотенціал
245
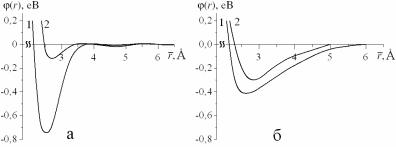
Хейне-Абаренкова-Анімалу [66]) з урахуванням різних обміннокореляційних поправок. Як можна бачити, криві 1 та 2 з різними об- мінно-кореляційними поправками відрізняються суттєво, це свідчить про невідповідності в розрахунках ефективних потенціалів міжатомної взаємодії в теорії псевдопотенціалів. Значно менша різниця в залежностях енергії взаємодії атомів від відстані (криві 1,2 на рис. 7.21,б) при розрахунках за МЗА в роботах різних авторів [85,86] свідчить про більш точність цього методу. Але при використанні для алюмінію моделі з вільною поверхнею [97] із жодним з цих потенціалів не були отримані вірні значення температури та теплоти плавлення.
Рис 7.21. Потенціали міжатомної взаємодії для Al;
а – ефективні потенціали для рідкого Al при T=970 К, розраховані за методом псевдопотенціалу з різними обмінно-кореляційними поправками, крива 1 – Шоу [87], 2 – Вашиста-Сингві [88]; б – потенціали взаємодії атомів розраховані за МЗА, крива 1 – [85], 2 – [86].
7.4.2. Основні вимоги до потенціалів, принципи їх вибору та корегування
Обчислення свідчать про те, що такі інтегральні характеристики як енергія зв’язку атомів, теплота випаровування, температури плавлення й поліморфного перетворення, ентальпія плавлення, пружні константи, модуль всебічного стиснення і т. д. сильно залежать від вибору потенціалу взаємодії. Останнім часом з’являється багато робіт, присвячених удосконаленню потенціалів таким чином, щоб вони відповідали більшій кількості фізичних характеристик, наприклад [89]. Однак отримати під час моделювання за методом МД задовільні значення достатньо великої кількості фізичних характеристик не вдається нікому.
246
Звісно, виникає проблема вибору потенціалу оптимальної версії. Відомо, що наявність асиметрії основної потенціальної ями забезпечує теплове розширення матеріалу. Теплота плавлення й теплота випаровування модельної речовини залежать від радіусу обрізання потенціалу, оскільки при цьому змінюється число сусідніх атомів, взаємодія з якими враховується. Цей фактор впливає і на міжатомну відстань, яка часто не відповідає положенню мінімуму на кривій потенціальної енергії. Температура плавлення і модулі пружності пов’язані в першу чергу з кривиною ходу потенціалу поблизу основного мінімуму. Характер осциляцій розрахованих потенціалів пов’язаний із кристалічною структурою. І навпаки, змінюючи розміщення осциляцій на вісі відстаней можна змоделювати різні кристалічні структури.
Процес плавлення призводить до розмиття поверхні Фермі атомів близько 15%, порівняно до кристалічного стану речовини. Осцилюючий характер ефективних потенціалів міжатомної взаємодії, притаманний розрахункам в теорії псевдопотенціалів, при переході від твердого тіла до рідини має ставати менш виразним (див. рис. 7.20 й рис. 7.21,а). Виникає питання можливості застосування потенціалів, розрахованих для кристалічного стану, речовини при моделюванні аморфних, некристалічних матеріалів. Основною проблемою застосування потенціалів є врахування змін електронної густини при змінах атомної структури та густини. У зв’язку з цим було розроблено узагальнений квантово-класичний метод Кара-Парінелло [90], в якому класичне трактування руху ядер, що розраховується методом МД, поєднується з квантовим описом руху електронів. В цьому методі потенціали міжатомної взаємодії не беруться параметрично, а розраховуються на кожному ітераційному кроці моделювання для кожного конкретного розташування атомів. Зрозуміло, що таке моделювання потребує чималих ресурсів (див. вище про метод ab-initio). Однак такі роботи вже відомі, наприклад [75,91].
7.4.3. Визначення потенціалів міжатомної взаємодії для змішаних систем
Великий інтерес для сучасного матеріалознавства становлять багатокомпонентні системи в аморфному або нанокристалічному стані, з притаманними їм фізико-хімічними властивостями. Наприклад, серед високоміцних алюмінієвих сплавів сьогодні особлива увага приділяється аморфним сплавам, що містять рідкоземельні елементи в комплексі з 3d металами. Ці нові сплави мають високі механічні
247
властивості, і для них характерні достатньо широкі діапазони термічної стабільності [92,93].
В силу важливості вивчення змішаних систем в багатьох роботах розробляються потенціали міжатомної взаємодії для атомів різних сортів. Співробітниками інституту металофізики ім. Г.В. Курдюмова НАН України проведено першопринципові розрахунки потенціалів міжатомної взаємодії багатокомпонентних систем з урахуванням кристалічних структур сполук, які утворюються в системах, що розглядаються. Наприклад, в роботі [72] для системи Al-Y-Nі потенціали парної взаємодії визначались на основі гіпотетичних кристалічних структур, які відповідали впорядкованим надструктурам заміщення атомами Y й Nі атомів Al у ГЦК гратці.
Але у більшості сучасних робіт автори використовують МЗА і його більш поліпшені модифікації. Наприклад, потенціали для системи Al-Ni [94] та для системи Al-Тi [88] розроблені на базі МЗА, а для системи Al-Mg [81] розрахунки проводились за ММЗА.
В роботі [94] в моделі МЗА сумарна енергія системи у випадку бінарних сплавів має вигляд
Etot = |
1 |
∑ϕai ,a j |
(rij ) + ∑Fai (ρi ), |
(7.59) |
|
||||
|
2 i, j |
i |
|
|
де ϕαi ,α j (rij ) – парний потенціал взаємодії як функція відстані rij між атомами i та j різного хімічного типу αi та αj (A й B); Fαi – енергія
занурення атома хімічного типу αi як функція локальної електронної густини ρi в точці i, індукованої усіма атомами системи. Локальна електронна густина ρi розраховується за співвідношенням
ρi = ∑ρa j (rij ) , |
(7.60) |
j¹i |
|
де ρaj (rij ) – функція електронної густини, визначена для атома хімі-
чного типу αj . Ця модель включає сім функцій взаємодій атомів, які можуть бути відповідно розділені на три групи:
1)ϕA,A , ρA (r) , FA (ρ) – для атомів сорту А і пар А-А;
2)ϕB,B , ρB (r) , FB (ρ) – для атомів сорту В і пар В-В;
3)ϕA,B – описують взаємодію атомів різного сорту (пар А-В).
248

Автори [81] на базі ММЗА розраховували енергію утворення сплаву AlMg з різними кристалічними структурами згідно з формулою
Hf = (Etot − NMgεMg − NAlεAl)/(NMg + NAl), |
(7.61) |
де Etot – повна енергія системи, NMg й NAl кількість атомів в системі
Mg й Al відповідно, εMg й εAl повна енергія на атом Mg й Al відповідно, яка розрахована для випадку ідеальної гратки відповідної фази, і порівняли отримані результати з результатами розрахунків у рамках ТФГ рис. 7.22.
З рис. 7.22 видно, що залежність теплоти утворення фази від об’єму елементарної комірки, розрахована за ММЗА, при порівнянні з результатами розрахунку за ТФГ, для випадку структури типу В1 узгоджується краще, ніж результати розрахунків для структур типу В2 й В3.
Рис. 7.22. Теплоти утворення кристалічних фаз сплаву AlMg,
В1, В2, В3 – структури на основі ОЦК, алмазної й ГЦК граток.
Найчастіше потенціали звіряються з експериментальними даними та з розрахунками із перших принципів для наступних властивостей: енергія як функція об’єму (загальна або в розрахунку на один атом) для різних кристалічних структур, теплоти утворення кристалічних фаз в двокомпонентних сплавах, об’єм, що приходиться на атом в рівноважному стані при заданій температурі (коефіцієнт об’ємного температурного розширення), пружні константи й модуль всебічного стиснення, температура й теплота плавлення, енергія утворення вакансій, енергії впровадження й заміщення атомів, фононний спектр.
249
7.5. Моделювання утворення аморфних та нанокристалічних матеріалів
7.5.1. Моделювання структуроутворення під час оскляння
Фундаментальна проблема перебудови атомної структури розплаву під час склоутворення залишається предметом численних досліджень протягом кількох десятиріч. В даний час одним з найбільш ефективних підходів до вивчення принципів структурної організації невпорядкованих систем є комп'ютерне моделювання.
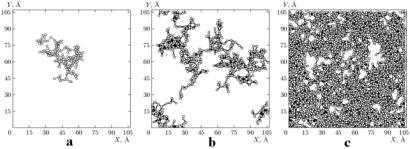
У [95,96] була запропонована структурна модель оскляння чистих металів, що встановлює принципові відмінності між атомною структурою розплаву і металевого скла. В рамках методу молекулярної динаміки на прикладі моделі було показано, що в основі структурної організації аморфної фази чистих металів лежить утворення з початком склоутворення і подальше зростання перколяційнного кластера (у лабораторній роботі № 14 у допоміжному посібнику описані перколяційні кластери та методи визначення їхньої фрактальної розмірності) із взаємопроникаючих і таких, що контактують між собою ікосаедрів, у вершинах і центрах яких розташовані атоми. Під взаємопроникаючими автори [95,96] розуміються ікосаедри (див. схему ікосаедра на рис. 7.18,д), у яких атоми, розміщені в центрах одних, одночасно є вершинами інших ікосаедрів. Ікосаедри, що контактують по граням і вершинам мають відповідно три і один загальний атом. Автори [96] вважають, що виявлені закономірності самоорганізації ікосаедрічної субструктури під час склоутворення корелюють з температурними залежностями основних термодинамічних характеристик моделі, які виявляють деякі ознаки фазового переходу другого роду і пояснюють їх поведінку на мікроскопічному рівні.
Рис. 7.23. Проекція на грань розрахункової комірки найбільшого кластера з контактуючих ікосаедрів [95],
для a,b,c температура Т=1460, 1260 і 1180 К.
250