

- •Contents
- •Contributors
- •Part I General Principles of Cell Death
- •1 Human Caspases – Apoptosis and Inflammation Signaling Proteases
- •1.1. Apoptosis and limited proteolysis
- •1.2. Caspase evolution
- •2. ACTIVATION MECHANISMS
- •2.2. The activation platforms
- •2.4. Proteolytic maturation
- •3. CASPASE SUBSTRATES
- •4. REGULATION BY NATURAL INHIBITORS
- •REFERENCES
- •2 Inhibitor of Apoptosis Proteins
- •2. CELLULAR FUNCTIONS AND PHENOTYPES OF IAP
- •3. IN VIVO FUNCTIONS OF IAP FAMILY PROTEINS
- •4. SUBCELLULAR LOCATIONS OF IAP
- •8. IAP–IAP INTERACTIONS
- •10. ENDOGENOUS ANTAGONISTS OF IAP
- •11. IAPs AND DISEASE
- •SUGGESTED READINGS
- •1. INTRODUCTION
- •2.1. The CD95 (Fas/APO-1) system
- •2.1.1. CD95 and CD95L: discovery of the first direct apoptosis-inducing receptor-ligand system
- •2.1.2. Biochemistry of CD95 apoptosis signaling
- •2.2. The TRAIL (Apo2L) system
- •3.1. The TNF system
- •3.1.1. Biochemistry of TNF signal transduction
- •3.1.2. TNF and TNF blockers in the clinic
- •3.2. The DR3 system
- •4. THE DR6 SYSTEM
- •6. CONCLUDING REMARKS AND OUTLOOK
- •SUGGESTED READINGS
- •4 Mitochondria and Cell Death
- •1. INTRODUCTION
- •2. MITOCHONDRIAL PHYSIOLOGY
- •3. THE MITOCHONDRIAL PATHWAY OF APOPTOSIS
- •9. CONCLUSIONS
- •SUGGESTED READINGS
- •1. INTRODUCTION
- •3. INHIBITING APOPTOSIS
- •4. INHIBITING THE INHIBITORS
- •6. THE BCL-2 FAMILY AND CANCER
- •SUGGESTED READINGS
- •6 Endoplasmic Reticulum Stress Response in Cell Death and Cell Survival
- •1. INTRODUCTION
- •2. THE ESR IN YEAST
- •3. THE ESR IN MAMMALS
- •4. THE ESR AND CELL DEATH
- •5. THE ESR IN DEVELOPMENT AND TISSUE HOMEOSTASIS
- •6. THE ESR IN HUMAN DISEASE
- •7. CONCLUSION
- •7 Autophagy – The Liaison between the Lysosomal System and Cell Death
- •1. INTRODUCTION
- •2. AUTOPHAGY
- •2.2. Physiologic functions of autophagy
- •2.3. Autophagy and human pathology
- •3. AUTOPHAGY AND CELL DEATH
- •3.1. Autophagy as anti–cell death mechanism
- •3.2. Autophagy as a cell death mechanism
- •3.3. Molecular players of the autophagy–cell death cross-talk
- •4. AUTOPHAGY, CELLULAR DEATH, AND CANCER
- •5. CONCLUDING REMARKS AND PENDING QUESTIONS
- •SUGGESTED READINGS
- •8 Cell Death in Response to Genotoxic Stress and DNA Damage
- •1. TYPES OF DNA DAMAGE AND REPAIR SYSTEMS
- •2. DNA DAMAGE RESPONSE
- •2.2. Transducers
- •2.3. Effectors
- •4. CHROMATIN MODIFICATIONS
- •5. CELL CYCLE CHECKPOINT REGULATION
- •6. WHEN REPAIR FAILS: SENESCENCE VERSUS APOPTOSIS
- •6.1. DNA damage response and the induction of apoptosis
- •6.2. p53-independent mechanisms of apoptosis
- •6.3. DNA damage response and senescence induction
- •7. DNA DAMAGE FROM OXIDATIVE STRESS
- •SUGGESTED READINGS
- •9 Ceramide and Lipid Mediators in Apoptosis
- •1. INTRODUCTION
- •3.1. Basic cell signaling often involves small molecules
- •3.2. Sphingolipids are cell-signaling molecules
- •3.2.1. Ceramide induces apoptosis
- •3.2.2. Ceramide accumulates during programmed cell death
- •3.2.3. Inhibition of ceramide production alters cell death signaling
- •4.1. Ceramide is generated through SM hydrolysis
- •4.3. aSMase can be activated independently of extracellular receptors to regulate apoptosis
- •4.4. Controversial aspects of the role of aSMase in apoptosis
- •4.5. De novo ceramide synthesis regulates programmed cell death
- •4.6. p53 and Bcl-2–like proteins are connected to de novo ceramide synthesis
- •4.7. The role and regulation of de novo synthesis in ceramide-mediated cell death is poorly understood
- •5. CONCLUDING REMARKS AND FUTURE DIRECTIONS
- •5.1. Who? (Which enzyme?)
- •5.2. What? (Which ceramide?)
- •5.3. Where? (Which compartment?)
- •5.4. When? (At what steps?)
- •5.5. How? (Through what mechanisms?)
- •5.6. What purpose?
- •6. SUMMARY
- •SUGGESTED READINGS
- •1. General Introduction
- •1.1. Cytotoxic lymphocytes and apoptosis
- •2. CYTOTOXIC GRANULES AND GRANULE EXOCYTOSIS
- •2.1. Synthesis and loading of the cytotoxic granule proteins into the secretory granules
- •2.2. The immunological synapse
- •2.3. Secretion of granule proteins
- •2.4. Uptake of proapoptotic proteins into the target cell
- •2.5. Activation of death pathways by granzymes
- •3. GRANULE-BOUND CYTOTOXIC PROTEINS
- •3.1. Perforin
- •3.2. Granulysin
- •3.3. Granzymes
- •3.3.1. GrB-mediated apoptosis
- •3.3.2. GrA-mediated cell death
- •3.3.3. Orphan granzyme-mediated cell death
- •5. CONCLUSIONS
- •REFERENCES
- •Part II Cell Death in Tissues and Organs
- •1.1. Death by trophic factor deprivation
- •1.2. Key molecules regulating neuronal apoptosis during development
- •1.2.1. Roles of caspases and Apaf-1 in neuronal cell death
- •1.2.2. Role of Bcl-2 family members in neuronal cell death
- •1.3. Signal transduction from neurotrophins and neurotrophin receptors
- •1.3.1. Signals for survival
- •1.3.2. Signals for death
- •2.1. Apoptosis in neurodegenerative diseases
- •2.1.4. Amyotrophic lateral sclerosis
- •2.2. Necrotic cell death in neurodegenerative diseases
- •2.2.1. Calpains
- •2.2.2. Cathepsins
- •3. CONCLUSIONS
- •ACKNOWLEDGMENT
- •SUGGESTED READINGS
- •ACKNOWLEDGMENT
- •SUGGESTED READINGS
- •1. INTRODUCTION
- •5. S-NITROSYLATION OF PARKIN
- •7. POTENTIAL TREATMENT OF EXCESSIVE NMDA-INDUCED Ca2+ INFLUX AND FREE RADICAL GENERATION
- •8. FUTURE THERAPEUTICS: NITROMEMANTINES
- •9. CONCLUSIONS
- •Acknowledgments
- •SUGGESTED READINGS
- •3. MITOCHONDRIAL PERMEABILITY TRANSITION ACTIVATED BY Ca2+ AND OXIDATIVE STRESS
- •4.1. Mitochondrial apoptotic pathways
- •4.2. Bcl-2 family proteins
- •4.3. Caspase-dependent apoptosis
- •4.4. Caspase-independent apoptosis
- •4.5. Calpains in ischemic neural cell death
- •5. SUMMARY
- •ACKNOWLEDGMENTS
- •SUGGESTED READINGS
- •1. INTRODUCTION
- •2. HISTORICAL ANTECEDENTS
- •7.1. Activation of p21 waf1/cip1: Targeting extrinsic and intrinsic pathways to death
- •8. CONCLUSION
- •ACKNOWLEDGMENTS
- •REFERENCES
- •16 Apoptosis and Homeostasis in the Eye
- •1.1. Lens
- •1.2. Retina
- •2. ROLE OF APOPTOSIS IN DISEASES OF THE EYE
- •2.1. Glaucoma
- •2.2. Age-related macular degeneration
- •4. APOPTOSIS AND OCULAR IMMUNE PRIVILEGE
- •5. CONCLUSIONS
- •SUGGESTED READINGS
- •17 Cell Death in the Inner Ear
- •3. THE COCHLEA IS THE HEARING ORGAN
- •3.1. Ototoxic hair cell death
- •3.2. Aminoglycoside-induced hair cell death
- •3.3. Cisplatin-induced hair cell death
- •3.4. Therapeutic strategies to prevent hair cell death
- •3.5. Challenges to studies of hair cell death
- •4. SPIRAL GANGLION NEURON DEATH
- •4.1. Neurotrophic support from sensory hair cells and supporting cells
- •4.2. Afferent activity from hair cells
- •4.3. Molecular manifestations of spiral ganglion neuron death
- •4.4. Therapeutic interventions to prevent SGN death
- •ACKNOWLEDGMENTS
- •SUGGESTED READINGS
- •18 Cell Death in the Olfactory System
- •1. Introduction
- •2. Anatomical Aspects
- •3. Life and Death in the Olfactory System
- •3.1. Olfactory epithelium
- •3.2. Olfactory bulb
- •REFERENCES
- •1. Introduction
- •3.1. Beta cell death in the development of T1D
- •3.2. Mechanisms of beta cell death in type 1 diabetes
- •3.2.1. Apoptosis signaling pathways downstream of death receptors and inflammatory cytokines
- •3.2.2. Oxidative stress
- •3.3. Mechanisms of beta cell death in type 2 diabetes
- •3.3.1. Glucolipitoxicity
- •3.3.2. Endoplasmic reticulum stress
- •5. SUMMARY
- •Acknowledgments
- •REFERENCES
- •20 Apoptosis in the Physiology and Diseases of the Respiratory Tract
- •1. APOPTOSIS IN LUNG DEVELOPMENT
- •2. APOPTOSIS IN LUNG PATHOPHYSIOLOGY
- •2.1. Apoptosis in pulmonary inflammation
- •2.2. Apoptosis in acute lung injury
- •2.3. Apoptosis in chronic obstructive pulmonary disease
- •2.4. Apoptosis in interstitial lung diseases
- •2.5. Apoptosis in pulmonary arterial hypertension
- •2.6. Apoptosis in lung cancer
- •SUGGESTED READINGS
- •21 Regulation of Cell Death in the Gastrointestinal Tract
- •1. INTRODUCTION
- •2. ESOPHAGUS
- •3. STOMACH
- •4. SMALL AND LARGE INTESTINE
- •5. LIVER
- •6. PANCREAS
- •7. SUMMARY AND CONCLUDING REMARKS
- •SUGGESTED READINGS
- •22 Apoptosis in the Kidney
- •1. NORMAL KIDNEY STRUCTURE AND FUNCTION
- •3. APOPTOSIS IN ADULT KIDNEY DISEASE
- •4. REGULATION OF APOPTOSIS IN KIDNEY CELLS
- •4.1. Survival factors
- •4.2. Lethal factors
- •4.2.1. TNF superfamily cytokines
- •4.2.2. Other cytokines
- •4.2.3. Glucose
- •4.2.4. Drugs and xenobiotics
- •4.2.5. Ischemia-reperfusion and sepsis
- •5. THERAPEUTIC APPROACHES
- •SUGGESTED READINGS
- •1. INTRODUCTION
- •2. APOPTOSIS IN THE NORMAL BREAST
- •2.1. Occurrence and role of apoptosis in the developing breast
- •2.2.2. Death ligands and death receptor pathway
- •2.2.4. LIF-STAT3 proapoptotic signaling
- •2.2.5. IGF survival signaling
- •2.2.6. Regulation by adhesion
- •2.2.7. PI3K/AKT pathway: molecular hub for survival signals
- •2.2.8. Downstream regulators of apoptosis: the BCL-2 family members
- •3. APOPTOSIS IN BREAST CANCER
- •3.1. Apoptosis in breast tumorigenesis and cancer progression
- •3.2. Molecular dysregulation of apoptosis in breast cancer
- •3.2.1. Altered expression of death ligands and their receptors in breast cancer
- •3.2.2. Deregulation of prosurvival growth factors and their receptors
- •3.2.3. Alterations in cell adhesion and resistance to anoikis
- •3.2.4. Enhanced activation of the PI3K/AKT pathway in breast cancer
- •3.2.5. p53 inactivation in breast cancer
- •3.2.6. Altered expression of BCL-2 family of proteins in breast cancer
- •5. CONCLUSION
- •SUGGESTED READINGS
- •1. INTRODUCTION
- •2. DETECTING CELL DEATH IN THE FEMALE GONADS
- •4. APOPTOSIS AND FEMALE REPRODUCTIVE AGING
- •6. CONCLUDING REMARKS
- •REFERENCES
- •25 Apoptotic Signaling in Male Germ Cells
- •1. INTRODUCTION
- •3.1. Murine models
- •3.2. Primate models
- •3.3. Pathways of caspase activation and apoptosis
- •3.4. Apoptotic signaling in male germ cells
- •5. P38 MITOGEN-ACTIVATED PROTEIN KINASE (MAPK) AND NITRIC OXIDE (NO)–MEDIATED INTRINSIC PATHWAY SIGNALING CONSTITUTES A CRITICAL COMPONENT OF APOPTOTIC SIGNALING IN MALE GERM CELLS AFTER HORMONE DEPRIVATION
- •11. CONCLUSIONS AND PERSPECTIVES
- •REFERENCES
- •26 Cell Death in the Cardiovascular System
- •1. INTRODUCTION
- •2. CELL DEATH IN THE VASCULATURE
- •2.1. Apoptosis in the developing blood vessels
- •2.2. Apoptosis in atherosclerosis
- •2.2.1. Vascular smooth muscle cells
- •2.2.2. Macrophages
- •2.2.3. Regulation of apoptosis in atherosclerosis
- •2.2.4. Necrosis and autophagy in atherosclerosis
- •3. CELL DEATH IN THE MYOCARDIUM
- •3.1. Cell death in myocardial infarction
- •3.1.1. Apoptosis in myocardial infarction
- •3.1.2. Necrosis in myocardial infarction
- •3.1.3. Autophagy in myocardial infarction
- •3.2. Cell death in heart failure
- •3.2.1. Apoptosis in heart failure
- •3.2.2. Necrosis in heart failure
- •3.2.3. Autophagy in heart failure
- •4. CONCLUDING REMARKS
- •ACKNOWLEDGMENTS
- •REFERENCES
- •27 Cell Death Regulation in Muscle
- •1. INTRODUCTION TO MUSCLE
- •1.1. Skeletal muscle adaptation to endurance training
- •1.2. Myonuclear domains
- •2. MITOCHONDRIALLY MEDIATED APOPTOSIS IN MUSCLE
- •2.1. Skeletal muscle apoptotic susceptibility
- •4. APOPTOSIS IN MUSCLE DURING AGING AND DISEASE
- •4.1. Aging
- •4.2. Type 2 diabetes mellitus
- •4.3. Cancer cachexia
- •4.4. Chronic heart failure
- •6. CONCLUSION
- •SUGGESTED READINGS
- •28 Cell Death in the Skin
- •1. INTRODUCTION
- •2. CELL DEATH IN SKIN HOMEOSTASIS
- •2.1. Cornification and apoptosis
- •2.2. Death receptors in the skin
- •3. CELL DEATH IN SKIN PATHOLOGY
- •3.1. Sunburn
- •3.2. Skin cancer
- •3.3. Necrolysis
- •3.4. Pemphigus
- •3.5. Eczema
- •3.6. Graft-versus-host disease
- •4. CONCLUDING REMARKS AND PERSPECTIVES
- •ACKNOWLEDGMENTS
- •SUGGESTED READINGS
- •29 Apoptosis and Cell Survival in the Immune System
- •2.1. Survival of early hematopoietic progenitors
- •2.2. Sizing of the T-cell population
- •2.2.1. Establishing central tolerance
- •2.2.2. Peripheral tolerance
- •2.2.3. Memory T cells
- •2.3. Control of apoptosis in B-cell development
- •2.3.1. Early B-cell development
- •2.3.2. Deletion of autoreactive B cells
- •2.3.3. Survival and death of activated B cells
- •3. IMPAIRED APOPTOSIS AND LEUKEMOGENESIS
- •4. CONCLUSIONS
- •ACKNOWLEDGMENTS
- •REFERENCES
- •30 Cell Death Regulation in the Hematopoietic System
- •1. INTRODUCTION
- •2. HEMATOPOIETIC STEM CELLS
- •4. ERYTHROPOIESIS
- •5. MEGAKARYOPOIESIS
- •6. GRANULOPOIESIS
- •7. MONOPOIESIS
- •8. CONCLUSION
- •ACKNOWLEDGMENTS
- •REFERENCES
- •31 Apoptotic Cell Death in Sepsis
- •1. INTRODUCTION
- •2. HOST INFLAMMATORY RESPONSE TO SEPSIS
- •3. CLINICAL OBSERVATIONS OF CELL DEATH IN SEPSIS
- •3.1. Sepsis-induced apoptosis
- •3.2. Necrotic cell death in sepsis
- •4.1. Central role of apoptosis in sepsis mortality: immune effector cells and gut epithelium
- •4.2. Apoptotic pathways in sepsis-induced immune cell death
- •4.3. Investigations implicating the extrinsic apoptotic pathway in sepsis
- •4.4. Investigations implicating the intrinsic apoptotic pathway in sepsis
- •5. THE EFFECT OF APOPTOSIS ON THE IMMUNE SYSTEM
- •5.1. Cellular effects of an increased apoptotic burdens
- •5.2. Network effects of selective loss of immune cell types
- •5.3. Studies of immunomodulation by apoptotic cells in other fields
- •7. CONCLUSION
- •REFERENCES
- •32 Host–Pathogen Interactions
- •1. INTRODUCTION
- •2. FROM THE PATHOGEN PERSPECTIVE
- •2.1. Commensals versus pathogens
- •2.2. Pathogen strategies to infect the host
- •3. HOST DEFENSE
- •3.1. Antimicrobial peptides
- •3.2. PRRs and inflammation
- •3.2.1. TLRs
- •3.2.2. NLRs
- •3.2.3. The Nod signalosome
- •3.2.4. The inflammasome
- •3.3. Cell death
- •3.3.1. Apoptosis and pathogen clearance
- •3.3.2. Pyroptosis
- •3.2.3. Caspase-independent cell death
- •3.2.4. Autophagy and autophagic cell death
- •4. CONCLUSIONS
- •REFERENCES
- •Part III Cell Death in Nonmammalian Organisms
- •1. PHENOTYPE AND ASSAYS OF YEAST APOPTOSIS
- •2.1. Pheromone-induced cell death
- •2.1.1. Colony growth
- •2.1.2. Killer-induced cell death
- •3. EXTERNAL STIMULI THAT INDUCE APOPTOSIS IN YEAST
- •4. THE GENETICS OF YEAST APOPTOSIS
- •5. PROGRAMMED AND ALTRUISTIC AGING
- •SUGGESTED READINGS
- •34 Caenorhabditis elegans and Apoptosis
- •1. Overview
- •2. KILLING
- •3. SPECIFICATION
- •4. EXECUTION
- •4.1. DNA degradation
- •4.2. Mitochondrial elimination
- •4.3. Engulfment
- •5. SUMMARY
- •SUGGESTED READINGS
- •35 Apoptotic Cell Death in Drosophila
- •2. DROSOPHILA CASPASES AND PROXIMAL REGULATORS
- •6. CLOSING COMMENTS
- •SUGGESTED READINGS
- •36 Analysis of Cell Death in Zebrafish
- •1. INTRODUCTION
- •2. WHY USE ZEBRAFISH TO STUDY CELL DEATH?
- •2.2. Molecular techniques to rapidly assess gene function in embryos
- •2.2.1. Studies of gene function using microinjections into early embryos
- •2.2.2. In situ hybridization and immunohistochemistry
- •2.3. Forward genetic screening
- •2.4. Drug and small-molecule screening
- •2.5. Transgenesis
- •2.6. Targeted knockouts
- •3.1. Intrinsic apoptosis
- •3.2. Extrinsic apoptosis
- •3.3. Chk-1 suppressed apoptosis
- •3.4. Anoikis
- •3.5. Autophagy
- •3.6. Necrosis
- •4. DEVELOPMENTAL CELL DEATH IN ZEBRAFISH EMBRYOS
- •5. THE P53 PATHWAY
- •6. PERSPECTIVES AND FUTURE DIRECTIONS
- •SUGGESTED READING

374 |
MAYA SALEH |
Life cycle
Enter
Disseminate
and exit
Colonize
Replicate
Persist
Figure 32-3. The life cycle of a pathogen. For a pathogen to survive it needs to invade the host, colonize the site of infection while resisting expulsion from the host, replicate, then exit to disseminate and find another host that would supply a new replication site and source of nutrients. Courtesy of Stanley Falkow.
or expression levels of flagellin when crossing epithelial cell barriers7; these cells express, on their basolateral surface, TLR5, a PRR that senses flagellin and guards against pathogen invasion.8,9
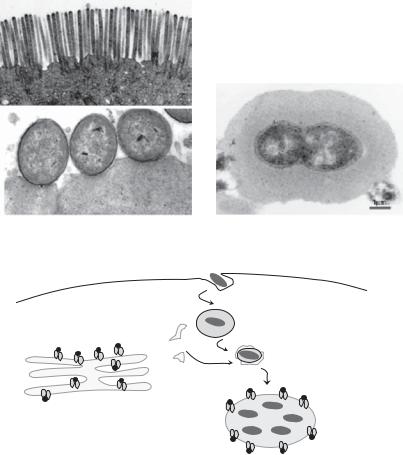
For intracellular pathogens, the challenge is of a different nature. They need to enter host cells, replicate without being recognized and degraded, and then exit the cell. A common strategy of intracellular pathogens is subversion of the endocytic machinery to their advantage and hiding in endosomal compartments. For instance, Brucella spp., as well as Legionella pneumophila, the causative agent of Legionnaire’s disease, surround their phagosomes with an endoplasmic reticulum–derived membrane that inhibits phagosomelysosome fusion, establishing an ideal niche for replication (Figure 32-4c).10 After replication, pathogens break out of their hiding places into the cytosol, where they induce cell death to exit the cell and disseminate. Pathogens secrete enzymes and toxins that allow their spread in tissues. Enteric pathogens induce death of intestinal epithelial cells or break their tight junctions to reach the submucosa. Various microbial toxins – such as nigericin, maitotoxin, aerolysin, gramicidin, pneumolysin, α-hemolysin, and α-toxin – form pores on the surface of host cells leading to their death. Some toxins derange the plasma membrane or the cytoskeleton, whereas others interfere with signal transduction pathways.11
3. HOST DEFENSE
inducible defense mechanisms. In mammals, constitutive defenses include the normal microbial flora that compete with pathogens, physical barriers of the skin and internal epithelial layers, mechanical defenses of the mucus and cilia, and chemical defenses such as the acidic pH of the stomach. For a long time, innate immunity was considered nonspecific. However, in the late 1980s, Janeway proposed that detection of pathogens by PRRs was key to specific activation of immune responses.12 Various classes of innate immunity recognition systems have been discovered. Some are soluble molecules such as mannose-binding lectins, ficolins, and collectins, whereas others are confined to cells, most notably, C-type lectins, Toll-like receptors (TLRs), nucleotide binding and oligomerization domain (Nod)– like receptors (NLRs), dsRNA helicase–like receptors (RIG-I and Mda5), and the cytoplasmic dsDNA receptors ZBP1-DAI and AIM2. PRRs recognize PAMP “signatures” displayed by microorganisms but not found on host cells. They also sense alterations in the host cellular environment arising indirectly from the infection, referred to as danger-associated molecular patterns (DAMPs), or alarm signals. Decrease in intracellular K+ levels, which occurs in response to pore-forming toxins, accumulation of reactive oxygen species (ROS), or release of “alarmins” in the extracellular space are examples of alarm signals or DAMPs that activate PRRs. Alarmins are unrelated host proteins with distinct cellular functions that acquire the ability to signal tissue damage and trigger an inflammatory response when secreted in the extracellular milieu in response to infection or cell death. Most notable are antimicrobial peptides, heat shock proteins, the non-histone chromatin-binding protein HMGB1, and extracellular matrix degradation products such as hyaluronan.13 Activation of PRRs is transduced via a plethora of cellular proteins (kinases, ubiquitin ligases, adaptors, proteases, transcription factors), which induce a large array of interconnected and synergistic defense mechanisms aimed at killing the pathogen while preserving host cell integrity. These defenses include the production of antimicrobial peptides by epithelial cells and neutrophils, elicitation of an inflammatory response necessary for the activation of phagocytes, and the death of infected cells that, in certain instances, limits the spread of pathogens to surrounding tissues.
3.1. Antimicrobial peptides
The host evolved means to detect pathogens’ intrusion and mechanisms to restrict their growth. Firstline defenses are provided by the innate immune system, which is armed by constitutive as well as
Antimicrobial peptides (AMPs) are endogenous antibiotics that have been established as an essential part of innate immunity. They bind to a wide variety of pathogens, including Gram-negative and Gram-positive
HOST–PATHOGEN INTERACTIONS |
375 |
(a) |
(b) |
Legionella
phagosome
Endoplasmic reticulum
 Ribosome
Ribosome
(c) Sec61 complex
Sec61 complex
bacteria, fungi, and some viruses, and disrupt their cytoplasmic membrane. In addition to their direct role in killing microbes, they act as immunostimulants that modulate the inflammatory response and chemoattract and activate antigen-presenting cells (APCs). They are small, generally cationic peptides with spaced hydrophobic and charged regions and are synthesized as prepropeptides with an N-terminal signal sequence, an anionic pro segment, and a C-terminal cationic AMP domain that gains biological activity after processing. In humans, AMPs are subgrouped into three classes based on structural characteristics: the defensins (α and β subfamilies), cathelicidins, and histatins (Figure 32-5).
Histatins (His-1 and His-3) are his- tidine-rich, mainly antifungal peptides found in the saliva. Defensins and cathelicidins are, on the other hand, expressed in neutrophils, keratinocytes, and epithelial cells either constitutively or on induction by bacteria and cytokines. More than 300 defensins have been identified so far in many organisms, including mammals, birds, invertebrates, and plants (http:// defensins.bii.a-star.edu.sg). In humans, there are six α-defensins. α-defensins 1 through 4, also known as human neutrophil peptides (HNP1–4), are stored as mature peptides in neutrophil azurophilic granules and contribute to nonoxidative killing of phagocytized pathogens. Human α-defensins 5 and 6 (HD-5 and -6) are primarily produced by epithelial cells and require processing upon release. In rodents, α-defensins are termed cryptdins, as they are mainly found in Paneth cells of the small intestinal crypts.
Cryptdins are processed |
by |
MMP7, |
a tissue metalloprotease |
also |
termed |
matrilysin; MMP7−/– mice lack mature cryptdins and are susceptible to oral infection with Salmonella.14 Unlike in mice, matrilysin is not found in the small intestine of humans; the digestive enzyme trypsin was found to be the cleaving enzyme for HD5.15 The question of why mice and humans use different enzymes to process Paneth cells defensins remains unclear. β- defensins (hBD1, hBD2, hBD3, and hBD4) are primarily produced by
epithelial cells. The processing of β-defensins is thought to occur in a similar fashion to that of α-defensins; however, the convertases involved remain unknown. α and β defensin subfamilies are characterized by three intramolecular disulfide bonds mediated by six conserved cysteine residues and differ by the cysteine pairing and the length of peptides between the cysteines (Figure 32-5). Structurally, they are folded in a characteristic three-stranded β sheet. θ-defensins, which form a third defensin subfamily, are structurally unrelated to α and β defensins and are only found in nonhuman primates. Experiments with genetically modified mice were most informative in confirming

MAYA SALEH
and synthetic θ-defensins (retrocyclins), were reported to neutralize the enzymatic activity of certain bacterial toxins, namely that of Bacillus anthracis’s lethal toxin (LT), diphtheria toxin (DT), and Pseudomonas endotoxin A (ETA), protecting from toxin-associated lethality both in vitro and in vivo.20 The only cathelicidin found in humans is LL-37/hCAP18. Its
murine ortholog cathelicidin-related AMP (CRAMP) has been shown in vivo to exert protective effects during bacterial infections.21 Consistent with previous observations suggesting anti-LPS activities by certain AMPs and protective effects from lethal endo-
toxemia, synthetic LL-37 derivatives
lacking bactericidal activity were shown to exert protective immunomodulatory activities in monocytes and macrophages. Specifically, one peptide termed innate defense regulator peptide (IDR-1) dampened the expression of proinflammatory cytokines in response to LPS while inducing chemokine levels and was efficacious in countering infections without obvious toxicities.22
In mammals, PRR signaling pathways, including those downstream of TLRs and NLRs, appear essential for the expression of AMPs, specifically β- defensins. In this aspect, there is a striking parallel in the regulation of AMP production between mammals and insects.23 Insects are notorious for
their resistance to infections. Their immune response depends heavily on the production of AMPs by the fat body, which is a functional equivalent of the mammalian liver. In Drosophila melanogaster, two distinct signaling pathways, referred to as Toll and Imd pathways, regulate AMP production. Within these pathways, three nuclear factor kappa B (NF-κB) proteins – namely Relish, Dorsal, and Dif – are central to the transcriptional induction of AMP genes. Fungi and Gram-positive bacteria largely activate the Toll pathway. Unlike mammalian TLRs, Drosophila Toll is not a PRR. It is activated by the cytokine spatzle,¨ which leads to the production of the antifungal AMP Drosomycin, which defends against fungal and Gram-positive bacterial infections. In contrast, the immune deficiency (Imd) pathway is