


CELL DEATH IN THE INNER EAR |
|
183 |
|
|
|
for sound transduction. Outer hair cell |
|
|
|
receptor potentials result in length |
|
|
|
changes of the outer hair cells along the |
|
|
|
length of their cell bodies. This “elec- |
|
|
|
tromotive force” serves to mechanically |
|
|
|
amplify sound energy transmission |
|
|
|
to the inner hair cells by 50 to 70 |
|
|
|
decibels. Outer hair cells are present |
|
|
|
only in mammalian inner ears, and |
|
|
|
they increase the range of frequencies |
|
|
|
that can be detected, as well as the |
|
|
|
frequency selectivity of the mammalian |
|
|
|
inner ear relative to that of birds or |
|
|
|
reptiles. In response to damaging |
|
|
|
stimuli such as noise or ototoxic drug |
|
|
|
exposure, outer hair cells are much |
|
Figure 17-1. The ear. The ear comprises three portions. The outer ear includes the external |
more susceptible to death than inner |
||
hair cells. |
|||
ear, ear canal, and the tympanic membrane. The middle ear is an air-filled space containing |
|||
the auditory ossicles (malleus, incus, and stapes). The inner ear is a fluid-filled space that |
Because of a mechanical compliance |
||
includes both the vestibular organs (three semicircular canals and the utricle and saccule |
gradient, the organ of Corti is arranged |
||
in the vestibule) and the hearing organ (cochlea). The vestibulocochlear nerve is divided |
|||
tonotopically such that the base of the |
|||
into the vestibular portion and the spiral ganglion portion, which innervates the cochlea. |
|||
Reprinted with permission from Encyclopaedia Britannica, C 1997 by Encyclopaedia Britan- |
cochlea detects high-frequency sounds, |
||
nica, Inc. See Color Plate 17. |
|
and lower frequency sounds are |
|
|
|
||
|
|
detected more apically (Figure 17-2). |
|
2. HAIR CELLS SYNAPSE WITH VESTIBULAR GANGLION |
The hair cells arranged along this tonotopic map are |
||
each responsive to a fairly narrow range of frequencies, |
|||
NEURONS AND SPIRAL GANGLION NEURONS |
|||
and each hair cell has a characteristic frequency at |
|||
|
|||
Hair cells in the six sensory patches of the inner ear |
which it is most sensitive. This frequency selectivity is |
||
make synaptic connections with bipolar first-order neu- |
largely a result of the properties of the basilar membrane |
||
rons of the eighth cranial nerve. The vestibular hair cells |
underlying the organ of Corti. This membrane vibrates |
||
are innervated by the vestibular ganglion portion of the |
in response to fluid motion in the cochlea. The basilar |
||
nerve, and the organ of Corti is innervated by the SGNs. |
membrane has gradients of both stiffness and width |
||
SGNs are primary auditory neurons that make synap- |
(narrow and stiff at the base; wide and compliant at |
||
tic connections with inner and outer hair cells of the |
the apex) that cause different regions of the membrane |
||
organ of Corti and thus serve as afferent neurons from |
to vibrate differentially according to the frequency of |
||
the peripheral organ of Corti to the cochlear nuclei in |
the stimulus. The ossicles in the middle ear transmit |
||
the central auditory system. In many cases, SGN survival |
sound energy into the fluid of the inner ear, driving the |
||
is intimately linked to the pathological status of sensory |
motion of the basilar membrane as a traveling wave |
||
hair cells, which provide trophic support for SGNs. |
that moves from base to apex and results in maximum |
||
|
basilar membrane motion at the region responsive to |
||
3. THE COCHLEA IS THE HEARING ORGAN |
the frequency of the stimulus. Because the traveling |
||
wave moves from base to apex, there is some displace- |
|||
|
|||
In the cochlea, two types of hair cells are contained |
ment of regions of the basilar membrane that are more |
||
within the organ of Corti. A single row of inner hair cells |
basal than the region maximally displaced, so there |
||
and three rows of outer hair cells run the length of the |
is also a lesser stimulation of higher-frequency hair |
||
organ of Corti from base to apex (Figure 17-2). Inner hair |
cells. Traumatic noise exposure can result in death |
||
cells are much more densely innervated than outer hair |
of hair cells that are most responsive to the frequen- |
||
cells. A single inner hair cell is innervated by ≥10 heav- |
cies contained in the traumatic stimulus. In addition, |
||
ily myelinated afferent nerve fibers, most of which are |
hair cells that are located more basal than the region |
||
contacted by a small efferent fiber. In contrast, a single |
of maximum basilar membrane displacement (i.e., |
||
unmyelinated afferent fiber will innervate many outer |
higher-frequency hair cells) can also be damaged or |
||
hair cells. Thus inner hair cells are primarily responsible |
killed. |
|
|
184 |
LISA L. CUNNINGHAM AND JUSTIN TAN |
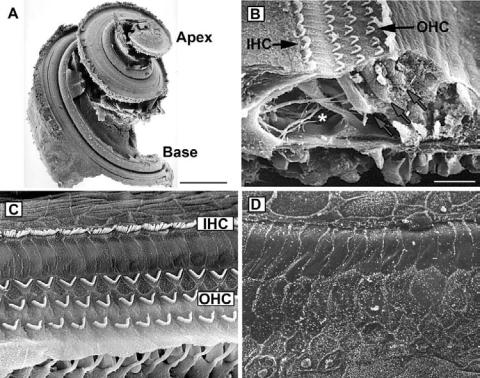
Figure 17-2. The cochlea. A. Scanning electron micrograph of an adult rat cochlea with the bony labyrinth dissected away. The cochlea spirals from base to apex, with sensory hair cells along the entire length. The base of the cochlea detects higher-frequency sounds, whereas the apex is more sensitive to low frequencies. B. Cross-section of a rat cochlea. Three rows of v-shaped outer hair cell (OHC) stereocilia bundles and a single row of inner hair cell (IHC) stereocilia can be seen at the upper center of the photo. Blue arrows point to outer hair cell bodies. Asterisk indicates the location of the tunnel of Corti, and green arrows point to e erent nerve processes (top arrow) and spiral ganglion fibers (lower arrow). C. Surface of the rat organ of Corti showing three rows of outer hair cell (OHC) stereocilia and a single row of inner hair cell (IHC) stereocilia. D. Cochlea of a rat treated with the aminoglycoside antibiotic amikacin. Both inner and outer hair cells at the base of the cochlea are missing. Scale bar in A = 2 mm. Scale bar in B = 20 μm. Figures reprinted with permission from Remy´ Pujol and Marc Lenoir, INSERM, France. See Color Plate 18.
3.1. Ototoxic hair cell death
Hair cells are sensitive to death from aging, noise trauma, and exposure to certain therapeutic drugs. Although this chapter focuses on mechanisms of hair cell death in response to ototoxic drug exposure, there is significant evidence that ototoxic hair cell death shares common molecular and signal transduction pathways with hair cell death caused by noise exposure and even aging. Two major classes of drugs have been identified that result in death of sensory hair cells in the inner ear. These ototoxic drugs are the aminoglycoside antibiotics and the antineoplastic agent cisplatin. Hair cell death in response to aminoglycosides and cisplatin will be discussed in more detail later in this chapter, but some important similarities exist between them in terms of their toxicity. First, both aminoglycosides and cisplatin are also nephrotoxic, and their ototoxic and nephrotoxic side effects are doselimiting for both drugs. Second, both aminoglycosides and cisplatin cause death of outer hair cells in the base of
the cochlea, resulting in a high-frequency sensory hearing loss. With continued exposure or increased doses, either aminoglycosides or cisplatin will result in progressively more apical (low-frequency) hair cell death. Both classes of drugs result in permanent hearing loss in a significant portion of humans receiving these drugs. Both drugs can also cause death of vestibular hair cells, resulting in permanent balance disorders.
3.2. Aminoglycoside-induced hair cell death
The aminoglycoside antibiotics were first discovered in the 1940s, and they remain among the most commonly used antibiotics worldwide. This class of antibiotics includes gentamicin, neomycin, amikacin, tobramycin, kanamycin, streptomycin, and paromomycin. Aminoglycosides are small (300–600 Dalton) molecules consisting of several saturated 6-carbon rings with attached amino groups. They have broad-spectrum bactericidal properties, especially against Gram-negative bacteria.
CELL DEATH IN THE INNER EAR |
185 |
Their primary antibacterial action is thought to be binding to the bacterial 16S ribosomal RNA and inhibiting protein translation. Aminoglycosides are very effective against Mycobacterium tuberculosis and are useful in the treatment of drug-resistant tuberculosis. They are also commonly used to treat Pseudomonas infections, including the recurrent Pseudomonas infections commonly seen in cystic fibrosis patients.
Shortly after their discovery, aminoglycosides were reported to have both ototoxic and nephrotoxic side effects. Sensory hair cells are the primary targets of aminoglycoside ototoxicity (Figure 17-2). Aminoglycosides are toxic to sensory hair cells in all species that have been examined, including aquatic vertebrates, reptiles, birds, and mammals. There is evidence that aminoglycosides enter sensory hair cells via the mechanotransduction channel itself. Once inside the hair cell, aminoglycosides accumulate in lysosomes. Transmission electron microscopic studies of hair cells treated with aminoglycosides have revealed early increases in the number of lysosomes, numerous vacuoles in the cytoplasm, and mitochondrial swelling and cristae disorganization. Scanning electron microscopic observations range from disorganization and fusion of stereocilia at low doses of aminoglycosides to complete loss of the hair cell at higher doses.
One proposed mechanism of aminoglycosideinduced hair cell death is the formation of reactive oxygen species (ROS) in the hair cell. Gentamicin has been shown to form a complex with iron that catalyzes the generation of free radicals, including superoxide, which can then lead to formation of the hydroxyl radical via iron-catalyzed Fenton reactions. The role of ROS formation in aminoglycoside-induced hair cell death is supported by evidence that ototoxicity is inhibited by both iron chelators and free radical scavengers, including glutathione.
Aminoglycoside-induced hair cell death has been characterized as apoptotic by both morphologic and molecular studies. Both cisplatin-induced and amino- glycoside-induced hair cell death are significantly inhibited by broad-spectrum inhibition of caspases. Experiments using fluorescent peptide caspase substrates have indicated that caspase-9 is a major mediator of aminoglycoside-induced hair cell death. Caspase-9 activation requires release of mitochondrial cytochrome c and apoptosome formation that results in autoactivation of caspase-9. Once activated, capase-9 activates caspase-3, a major executioner caspase that carries out the apoptotic program by cleaving proteins essential for cellular survival. Experiments using small-molecule inhibitors of X-linked inhibitor of apoptosis protein
(XIAP) suggest that XIAP may inhibit gentamicininduced activation of caspase-3 in hair cells. Overexpression of Bcl-2 also inhibits aminoglycoside-induced hair cell death and caspase-9 activation.
Aminoglycoside-induced hair cell death is mediated by activation of c-Jun NH2-terminal kinases (JNKs). JNKs are mitogen-activated protein kinases (MAPKs) that are activated in response to a variety of stresses, including inflammatory cytokines, osmotic stress, radiation, and excitotoxicity. Aminoglycosides result in phosphorylation of JNKs in hair cells. Inhibition of JNK signaling using a variety of approaches can protect hair cells against aminoglycoside toxicity. Activation of JNK is upstream of caspase-9 activation in hair cells. Interestingly, JNK inhibition does not protect against cisplatin-induced hair cell death, indicating that although aminoglycosideand cisplatininduced ototoxicity share common downstream molecular mechanisms, their upstream signaling mechanisms diverge.
3.3. Cisplatin-induced hair cell death
Cisplatin is among the most effective and widely used chemotherapeutic drugs available, and it is used worldwide to treat a broad range of tumors, including testicular, bladder, lung, stomach, and ovarian cancers. Estimates of the incidence of hearing loss in patients receiving cisplatin range from 12% to 90%, depending on the diagnosis, total dose of cisplatin received, and population examined. Cisplatin exposure results in damage or death of several cell types in the inner ear, including auditory and vestibular hair cells, stria vascularis, and spiral ganglion cells.
In cancer cells, cisplatin binds to DNA and results in DNA cross-linking that induces apoptosis via both p53-dependent and p53-independent mechanisms. The tumor suppressor p53 mediates cellular responses to DNA damage via transcriptional upregulation of genes that regulate cell cycle checkpoints as well as apoptosis. In addition, p53 can regulate cell fate via transcriptionindependent mechanisms. For example, p53 may promote cytochrome c release both indirectly by promoting the translocation of the proapoptotic Bcl-2 family protein Bax to the mitochondria and directly by translocating to mitochondria under apoptotic conditions. Studies in the auditory system have revealed that platinated DNA is present in most cells of the organ of Corti as well as in the stria vascularis in guinea pigs after exposure to cisplatin. One study reported that the p53 inhibitor pifithrin alpha inhibits cisplatin-induced hair cell death and caspase activation.