

- •Contents
- •Contributors
- •Part I General Principles of Cell Death
- •1 Human Caspases – Apoptosis and Inflammation Signaling Proteases
- •1.1. Apoptosis and limited proteolysis
- •1.2. Caspase evolution
- •2. ACTIVATION MECHANISMS
- •2.2. The activation platforms
- •2.4. Proteolytic maturation
- •3. CASPASE SUBSTRATES
- •4. REGULATION BY NATURAL INHIBITORS
- •REFERENCES
- •2 Inhibitor of Apoptosis Proteins
- •2. CELLULAR FUNCTIONS AND PHENOTYPES OF IAP
- •3. IN VIVO FUNCTIONS OF IAP FAMILY PROTEINS
- •4. SUBCELLULAR LOCATIONS OF IAP
- •8. IAP–IAP INTERACTIONS
- •10. ENDOGENOUS ANTAGONISTS OF IAP
- •11. IAPs AND DISEASE
- •SUGGESTED READINGS
- •1. INTRODUCTION
- •2.1. The CD95 (Fas/APO-1) system
- •2.1.1. CD95 and CD95L: discovery of the first direct apoptosis-inducing receptor-ligand system
- •2.1.2. Biochemistry of CD95 apoptosis signaling
- •2.2. The TRAIL (Apo2L) system
- •3.1. The TNF system
- •3.1.1. Biochemistry of TNF signal transduction
- •3.1.2. TNF and TNF blockers in the clinic
- •3.2. The DR3 system
- •4. THE DR6 SYSTEM
- •6. CONCLUDING REMARKS AND OUTLOOK
- •SUGGESTED READINGS
- •4 Mitochondria and Cell Death
- •1. INTRODUCTION
- •2. MITOCHONDRIAL PHYSIOLOGY
- •3. THE MITOCHONDRIAL PATHWAY OF APOPTOSIS
- •9. CONCLUSIONS
- •SUGGESTED READINGS
- •1. INTRODUCTION
- •3. INHIBITING APOPTOSIS
- •4. INHIBITING THE INHIBITORS
- •6. THE BCL-2 FAMILY AND CANCER
- •SUGGESTED READINGS
- •6 Endoplasmic Reticulum Stress Response in Cell Death and Cell Survival
- •1. INTRODUCTION
- •2. THE ESR IN YEAST
- •3. THE ESR IN MAMMALS
- •4. THE ESR AND CELL DEATH
- •5. THE ESR IN DEVELOPMENT AND TISSUE HOMEOSTASIS
- •6. THE ESR IN HUMAN DISEASE
- •7. CONCLUSION
- •7 Autophagy – The Liaison between the Lysosomal System and Cell Death
- •1. INTRODUCTION
- •2. AUTOPHAGY
- •2.2. Physiologic functions of autophagy
- •2.3. Autophagy and human pathology
- •3. AUTOPHAGY AND CELL DEATH
- •3.1. Autophagy as anti–cell death mechanism
- •3.2. Autophagy as a cell death mechanism
- •3.3. Molecular players of the autophagy–cell death cross-talk
- •4. AUTOPHAGY, CELLULAR DEATH, AND CANCER
- •5. CONCLUDING REMARKS AND PENDING QUESTIONS
- •SUGGESTED READINGS
- •8 Cell Death in Response to Genotoxic Stress and DNA Damage
- •1. TYPES OF DNA DAMAGE AND REPAIR SYSTEMS
- •2. DNA DAMAGE RESPONSE
- •2.2. Transducers
- •2.3. Effectors
- •4. CHROMATIN MODIFICATIONS
- •5. CELL CYCLE CHECKPOINT REGULATION
- •6. WHEN REPAIR FAILS: SENESCENCE VERSUS APOPTOSIS
- •6.1. DNA damage response and the induction of apoptosis
- •6.2. p53-independent mechanisms of apoptosis
- •6.3. DNA damage response and senescence induction
- •7. DNA DAMAGE FROM OXIDATIVE STRESS
- •SUGGESTED READINGS
- •9 Ceramide and Lipid Mediators in Apoptosis
- •1. INTRODUCTION
- •3.1. Basic cell signaling often involves small molecules
- •3.2. Sphingolipids are cell-signaling molecules
- •3.2.1. Ceramide induces apoptosis
- •3.2.2. Ceramide accumulates during programmed cell death
- •3.2.3. Inhibition of ceramide production alters cell death signaling
- •4.1. Ceramide is generated through SM hydrolysis
- •4.3. aSMase can be activated independently of extracellular receptors to regulate apoptosis
- •4.4. Controversial aspects of the role of aSMase in apoptosis
- •4.5. De novo ceramide synthesis regulates programmed cell death
- •4.6. p53 and Bcl-2–like proteins are connected to de novo ceramide synthesis
- •4.7. The role and regulation of de novo synthesis in ceramide-mediated cell death is poorly understood
- •5. CONCLUDING REMARKS AND FUTURE DIRECTIONS
- •5.1. Who? (Which enzyme?)
- •5.2. What? (Which ceramide?)
- •5.3. Where? (Which compartment?)
- •5.4. When? (At what steps?)
- •5.5. How? (Through what mechanisms?)
- •5.6. What purpose?
- •6. SUMMARY
- •SUGGESTED READINGS
- •1. General Introduction
- •1.1. Cytotoxic lymphocytes and apoptosis
- •2. CYTOTOXIC GRANULES AND GRANULE EXOCYTOSIS
- •2.1. Synthesis and loading of the cytotoxic granule proteins into the secretory granules
- •2.2. The immunological synapse
- •2.3. Secretion of granule proteins
- •2.4. Uptake of proapoptotic proteins into the target cell
- •2.5. Activation of death pathways by granzymes
- •3. GRANULE-BOUND CYTOTOXIC PROTEINS
- •3.1. Perforin
- •3.2. Granulysin
- •3.3. Granzymes
- •3.3.1. GrB-mediated apoptosis
- •3.3.2. GrA-mediated cell death
- •3.3.3. Orphan granzyme-mediated cell death
- •5. CONCLUSIONS
- •REFERENCES
- •Part II Cell Death in Tissues and Organs
- •1.1. Death by trophic factor deprivation
- •1.2. Key molecules regulating neuronal apoptosis during development
- •1.2.1. Roles of caspases and Apaf-1 in neuronal cell death
- •1.2.2. Role of Bcl-2 family members in neuronal cell death
- •1.3. Signal transduction from neurotrophins and neurotrophin receptors
- •1.3.1. Signals for survival
- •1.3.2. Signals for death
- •2.1. Apoptosis in neurodegenerative diseases
- •2.1.4. Amyotrophic lateral sclerosis
- •2.2. Necrotic cell death in neurodegenerative diseases
- •2.2.1. Calpains
- •2.2.2. Cathepsins
- •3. CONCLUSIONS
- •ACKNOWLEDGMENT
- •SUGGESTED READINGS
- •ACKNOWLEDGMENT
- •SUGGESTED READINGS
- •1. INTRODUCTION
- •5. S-NITROSYLATION OF PARKIN
- •7. POTENTIAL TREATMENT OF EXCESSIVE NMDA-INDUCED Ca2+ INFLUX AND FREE RADICAL GENERATION
- •8. FUTURE THERAPEUTICS: NITROMEMANTINES
- •9. CONCLUSIONS
- •Acknowledgments
- •SUGGESTED READINGS
- •3. MITOCHONDRIAL PERMEABILITY TRANSITION ACTIVATED BY Ca2+ AND OXIDATIVE STRESS
- •4.1. Mitochondrial apoptotic pathways
- •4.2. Bcl-2 family proteins
- •4.3. Caspase-dependent apoptosis
- •4.4. Caspase-independent apoptosis
- •4.5. Calpains in ischemic neural cell death
- •5. SUMMARY
- •ACKNOWLEDGMENTS
- •SUGGESTED READINGS
- •1. INTRODUCTION
- •2. HISTORICAL ANTECEDENTS
- •7.1. Activation of p21 waf1/cip1: Targeting extrinsic and intrinsic pathways to death
- •8. CONCLUSION
- •ACKNOWLEDGMENTS
- •REFERENCES
- •16 Apoptosis and Homeostasis in the Eye
- •1.1. Lens
- •1.2. Retina
- •2. ROLE OF APOPTOSIS IN DISEASES OF THE EYE
- •2.1. Glaucoma
- •2.2. Age-related macular degeneration
- •4. APOPTOSIS AND OCULAR IMMUNE PRIVILEGE
- •5. CONCLUSIONS
- •SUGGESTED READINGS
- •17 Cell Death in the Inner Ear
- •3. THE COCHLEA IS THE HEARING ORGAN
- •3.1. Ototoxic hair cell death
- •3.2. Aminoglycoside-induced hair cell death
- •3.3. Cisplatin-induced hair cell death
- •3.4. Therapeutic strategies to prevent hair cell death
- •3.5. Challenges to studies of hair cell death
- •4. SPIRAL GANGLION NEURON DEATH
- •4.1. Neurotrophic support from sensory hair cells and supporting cells
- •4.2. Afferent activity from hair cells
- •4.3. Molecular manifestations of spiral ganglion neuron death
- •4.4. Therapeutic interventions to prevent SGN death
- •ACKNOWLEDGMENTS
- •SUGGESTED READINGS
- •18 Cell Death in the Olfactory System
- •1. Introduction
- •2. Anatomical Aspects
- •3. Life and Death in the Olfactory System
- •3.1. Olfactory epithelium
- •3.2. Olfactory bulb
- •REFERENCES
- •1. Introduction
- •3.1. Beta cell death in the development of T1D
- •3.2. Mechanisms of beta cell death in type 1 diabetes
- •3.2.1. Apoptosis signaling pathways downstream of death receptors and inflammatory cytokines
- •3.2.2. Oxidative stress
- •3.3. Mechanisms of beta cell death in type 2 diabetes
- •3.3.1. Glucolipitoxicity
- •3.3.2. Endoplasmic reticulum stress
- •5. SUMMARY
- •Acknowledgments
- •REFERENCES
- •20 Apoptosis in the Physiology and Diseases of the Respiratory Tract
- •1. APOPTOSIS IN LUNG DEVELOPMENT
- •2. APOPTOSIS IN LUNG PATHOPHYSIOLOGY
- •2.1. Apoptosis in pulmonary inflammation
- •2.2. Apoptosis in acute lung injury
- •2.3. Apoptosis in chronic obstructive pulmonary disease
- •2.4. Apoptosis in interstitial lung diseases
- •2.5. Apoptosis in pulmonary arterial hypertension
- •2.6. Apoptosis in lung cancer
- •SUGGESTED READINGS
- •21 Regulation of Cell Death in the Gastrointestinal Tract
- •1. INTRODUCTION
- •2. ESOPHAGUS
- •3. STOMACH
- •4. SMALL AND LARGE INTESTINE
- •5. LIVER
- •6. PANCREAS
- •7. SUMMARY AND CONCLUDING REMARKS
- •SUGGESTED READINGS
- •22 Apoptosis in the Kidney
- •1. NORMAL KIDNEY STRUCTURE AND FUNCTION
- •3. APOPTOSIS IN ADULT KIDNEY DISEASE
- •4. REGULATION OF APOPTOSIS IN KIDNEY CELLS
- •4.1. Survival factors
- •4.2. Lethal factors
- •4.2.1. TNF superfamily cytokines
- •4.2.2. Other cytokines
- •4.2.3. Glucose
- •4.2.4. Drugs and xenobiotics
- •4.2.5. Ischemia-reperfusion and sepsis
- •5. THERAPEUTIC APPROACHES
- •SUGGESTED READINGS
- •1. INTRODUCTION
- •2. APOPTOSIS IN THE NORMAL BREAST
- •2.1. Occurrence and role of apoptosis in the developing breast
- •2.2.2. Death ligands and death receptor pathway
- •2.2.4. LIF-STAT3 proapoptotic signaling
- •2.2.5. IGF survival signaling
- •2.2.6. Regulation by adhesion
- •2.2.7. PI3K/AKT pathway: molecular hub for survival signals
- •2.2.8. Downstream regulators of apoptosis: the BCL-2 family members
- •3. APOPTOSIS IN BREAST CANCER
- •3.1. Apoptosis in breast tumorigenesis and cancer progression
- •3.2. Molecular dysregulation of apoptosis in breast cancer
- •3.2.1. Altered expression of death ligands and their receptors in breast cancer
- •3.2.2. Deregulation of prosurvival growth factors and their receptors
- •3.2.3. Alterations in cell adhesion and resistance to anoikis
- •3.2.4. Enhanced activation of the PI3K/AKT pathway in breast cancer
- •3.2.5. p53 inactivation in breast cancer
- •3.2.6. Altered expression of BCL-2 family of proteins in breast cancer
- •5. CONCLUSION
- •SUGGESTED READINGS
- •1. INTRODUCTION
- •2. DETECTING CELL DEATH IN THE FEMALE GONADS
- •4. APOPTOSIS AND FEMALE REPRODUCTIVE AGING
- •6. CONCLUDING REMARKS
- •REFERENCES
- •25 Apoptotic Signaling in Male Germ Cells
- •1. INTRODUCTION
- •3.1. Murine models
- •3.2. Primate models
- •3.3. Pathways of caspase activation and apoptosis
- •3.4. Apoptotic signaling in male germ cells
- •5. P38 MITOGEN-ACTIVATED PROTEIN KINASE (MAPK) AND NITRIC OXIDE (NO)–MEDIATED INTRINSIC PATHWAY SIGNALING CONSTITUTES A CRITICAL COMPONENT OF APOPTOTIC SIGNALING IN MALE GERM CELLS AFTER HORMONE DEPRIVATION
- •11. CONCLUSIONS AND PERSPECTIVES
- •REFERENCES
- •26 Cell Death in the Cardiovascular System
- •1. INTRODUCTION
- •2. CELL DEATH IN THE VASCULATURE
- •2.1. Apoptosis in the developing blood vessels
- •2.2. Apoptosis in atherosclerosis
- •2.2.1. Vascular smooth muscle cells
- •2.2.2. Macrophages
- •2.2.3. Regulation of apoptosis in atherosclerosis
- •2.2.4. Necrosis and autophagy in atherosclerosis
- •3. CELL DEATH IN THE MYOCARDIUM
- •3.1. Cell death in myocardial infarction
- •3.1.1. Apoptosis in myocardial infarction
- •3.1.2. Necrosis in myocardial infarction
- •3.1.3. Autophagy in myocardial infarction
- •3.2. Cell death in heart failure
- •3.2.1. Apoptosis in heart failure
- •3.2.2. Necrosis in heart failure
- •3.2.3. Autophagy in heart failure
- •4. CONCLUDING REMARKS
- •ACKNOWLEDGMENTS
- •REFERENCES
- •27 Cell Death Regulation in Muscle
- •1. INTRODUCTION TO MUSCLE
- •1.1. Skeletal muscle adaptation to endurance training
- •1.2. Myonuclear domains
- •2. MITOCHONDRIALLY MEDIATED APOPTOSIS IN MUSCLE
- •2.1. Skeletal muscle apoptotic susceptibility
- •4. APOPTOSIS IN MUSCLE DURING AGING AND DISEASE
- •4.1. Aging
- •4.2. Type 2 diabetes mellitus
- •4.3. Cancer cachexia
- •4.4. Chronic heart failure
- •6. CONCLUSION
- •SUGGESTED READINGS
- •28 Cell Death in the Skin
- •1. INTRODUCTION
- •2. CELL DEATH IN SKIN HOMEOSTASIS
- •2.1. Cornification and apoptosis
- •2.2. Death receptors in the skin
- •3. CELL DEATH IN SKIN PATHOLOGY
- •3.1. Sunburn
- •3.2. Skin cancer
- •3.3. Necrolysis
- •3.4. Pemphigus
- •3.5. Eczema
- •3.6. Graft-versus-host disease
- •4. CONCLUDING REMARKS AND PERSPECTIVES
- •ACKNOWLEDGMENTS
- •SUGGESTED READINGS
- •29 Apoptosis and Cell Survival in the Immune System
- •2.1. Survival of early hematopoietic progenitors
- •2.2. Sizing of the T-cell population
- •2.2.1. Establishing central tolerance
- •2.2.2. Peripheral tolerance
- •2.2.3. Memory T cells
- •2.3. Control of apoptosis in B-cell development
- •2.3.1. Early B-cell development
- •2.3.2. Deletion of autoreactive B cells
- •2.3.3. Survival and death of activated B cells
- •3. IMPAIRED APOPTOSIS AND LEUKEMOGENESIS
- •4. CONCLUSIONS
- •ACKNOWLEDGMENTS
- •REFERENCES
- •30 Cell Death Regulation in the Hematopoietic System
- •1. INTRODUCTION
- •2. HEMATOPOIETIC STEM CELLS
- •4. ERYTHROPOIESIS
- •5. MEGAKARYOPOIESIS
- •6. GRANULOPOIESIS
- •7. MONOPOIESIS
- •8. CONCLUSION
- •ACKNOWLEDGMENTS
- •REFERENCES
- •31 Apoptotic Cell Death in Sepsis
- •1. INTRODUCTION
- •2. HOST INFLAMMATORY RESPONSE TO SEPSIS
- •3. CLINICAL OBSERVATIONS OF CELL DEATH IN SEPSIS
- •3.1. Sepsis-induced apoptosis
- •3.2. Necrotic cell death in sepsis
- •4.1. Central role of apoptosis in sepsis mortality: immune effector cells and gut epithelium
- •4.2. Apoptotic pathways in sepsis-induced immune cell death
- •4.3. Investigations implicating the extrinsic apoptotic pathway in sepsis
- •4.4. Investigations implicating the intrinsic apoptotic pathway in sepsis
- •5. THE EFFECT OF APOPTOSIS ON THE IMMUNE SYSTEM
- •5.1. Cellular effects of an increased apoptotic burdens
- •5.2. Network effects of selective loss of immune cell types
- •5.3. Studies of immunomodulation by apoptotic cells in other fields
- •7. CONCLUSION
- •REFERENCES
- •32 Host–Pathogen Interactions
- •1. INTRODUCTION
- •2. FROM THE PATHOGEN PERSPECTIVE
- •2.1. Commensals versus pathogens
- •2.2. Pathogen strategies to infect the host
- •3. HOST DEFENSE
- •3.1. Antimicrobial peptides
- •3.2. PRRs and inflammation
- •3.2.1. TLRs
- •3.2.2. NLRs
- •3.2.3. The Nod signalosome
- •3.2.4. The inflammasome
- •3.3. Cell death
- •3.3.1. Apoptosis and pathogen clearance
- •3.3.2. Pyroptosis
- •3.2.3. Caspase-independent cell death
- •3.2.4. Autophagy and autophagic cell death
- •4. CONCLUSIONS
- •REFERENCES
- •Part III Cell Death in Nonmammalian Organisms
- •1. PHENOTYPE AND ASSAYS OF YEAST APOPTOSIS
- •2.1. Pheromone-induced cell death
- •2.1.1. Colony growth
- •2.1.2. Killer-induced cell death
- •3. EXTERNAL STIMULI THAT INDUCE APOPTOSIS IN YEAST
- •4. THE GENETICS OF YEAST APOPTOSIS
- •5. PROGRAMMED AND ALTRUISTIC AGING
- •SUGGESTED READINGS
- •34 Caenorhabditis elegans and Apoptosis
- •1. Overview
- •2. KILLING
- •3. SPECIFICATION
- •4. EXECUTION
- •4.1. DNA degradation
- •4.2. Mitochondrial elimination
- •4.3. Engulfment
- •5. SUMMARY
- •SUGGESTED READINGS
- •35 Apoptotic Cell Death in Drosophila
- •2. DROSOPHILA CASPASES AND PROXIMAL REGULATORS
- •6. CLOSING COMMENTS
- •SUGGESTED READINGS
- •36 Analysis of Cell Death in Zebrafish
- •1. INTRODUCTION
- •2. WHY USE ZEBRAFISH TO STUDY CELL DEATH?
- •2.2. Molecular techniques to rapidly assess gene function in embryos
- •2.2.1. Studies of gene function using microinjections into early embryos
- •2.2.2. In situ hybridization and immunohistochemistry
- •2.3. Forward genetic screening
- •2.4. Drug and small-molecule screening
- •2.5. Transgenesis
- •2.6. Targeted knockouts
- •3.1. Intrinsic apoptosis
- •3.2. Extrinsic apoptosis
- •3.3. Chk-1 suppressed apoptosis
- •3.4. Anoikis
- •3.5. Autophagy
- •3.6. Necrosis
- •4. DEVELOPMENTAL CELL DEATH IN ZEBRAFISH EMBRYOS
- •5. THE P53 PATHWAY
- •6. PERSPECTIVES AND FUTURE DIRECTIONS
- •SUGGESTED READING
34 Caenorhabditis elegans and Apoptosis
Brian L. Harry and Ding Xue
One point that emerges from the studies of programmed cell death in C. elegans and other organisms is the striking similarity of genes and gene pathways among organisms that are as superficially distinct as worms and humans.
– H. Robert Horvitz, 2002 Nobel Lecture
1. OVERVIEW
Since Caenorhabditis elegans was pioneered as a research tool in the early 1970s, it has become a window for peering into programmed cell death (PCD), also known as apoptosis. A soil-dwelling organism that is 1 mm in length and transparent, C. elegans is well suited for microscopic inquiry. Its reproductive cycle lasts 2 to 3 days and begins with either self-fertilization of a hermaphrodite or cross-fertilization between a hermaphrodite and a male. Forward genetic approaches using random mutagenesis and phenotypic screening, combined with a rapid generation turnover, have made C. elegans a powerful platform for discovery of new genes in PCD.
Cells in C. elegans can be tracked throughout development from their precursors because of a remarkably invariant cell lineage, which enables study of PCD at single-cell resolution. Of the 1,090 somatic cells that are generated during the life of a C. elegans hermaphrodite, 113 undergo PCD during embryogenesis and 18 during early larval development. These cell deaths are consistent and predictable according to their cell lineage. A third wave of PCD in the germline of adult worms occurs in a stochastic, lineage-independent manner. It has been estimated that roughly half of the 2,000 germ cells produced during a worm’s lifespan undergo apoptosis. Apoptotic cells can be recognized by their highly refractile, raised disc morphology under differential interference contrast (DIC) microscopy (Figure 34-1). These unique features have made C. elegans an excellent model for elucidating fundamental aspects of apoptosis.
Apoptosis in multicellular eukaryotic systems can be cued by extrinsic signals that act through surface receptors or by intrinsic signals that trigger changes in mitochondria. Developmental PCD in C. elegans occurs
predominantly through the intrinsic pathway, also known as cell-autonomous death. Intrinsic signals initiate the specification phase of PCD, which decides the life versus death fate of a cell. Death specification pathways differ among cells, but they converge on a few targets, in particular, the cell death initiator egl-1 (egl, egg-laying defective). In many cases, transcriptional upregulation of egl-1 signifies the killing phase, which culminates in the activation of an aspartate-specific cysteine protease encoded by the ced-3 gene (ced, cell death abnormal). The activated CED-3 protease then initiates the execution phase through cleavage and activation of downstream effectors that promote chromosomal degradation, mitochondrial elimination, alteration of the cell membrane, and removal of the dying cells. We discuss each of these phases for somatic PCD, beginning with the killing phase.
2. KILLING
A core genetic pathway is responsible for almost all PCD events during C. elegans development. Loss-of-function (lf ) mutations in egl-1, ced-4, and ced-3 result in survival of cells destined to die. Over-expression or increased function of these genes promotes ectopic cell deaths. On the other hand, ced-9 (lf ) mutations result in excessive cell death and lethality, whereas a gain-of-function (gf ) mutation in ced-9 prevents cell death. Epistasis analysis suggests that egl-1 acts upstream of ced-9 to induce apoptosis, ced-9 acts upstream of ced-4 to prevent apoptosis, and ced-4 acts upstream of ced-3 to induce apoptosis (Figure 34-2). When egl-1 transcription is upregulated, the result of this pathway is activation of the CED-3 protease, which is a member of the aspartatespecific cysteine proteases, also known as caspases, that are involved in apoptosis in diverse species.
397

398 |
BRIAN L. HARRY AND DING XUE |
CED-3 cleaves its substrates at aspartate residues and is responsible for activating enzymes that degrade chromosomes, eliminate mitochondria, and promote cell surface changes to permit recognition by an engulfing cell (see Section 4, Execution).
Biochemical and structural experiments have corroborated the preceding genetic model and elucidated the functions of these proteins. In the early 1990s, CED-9 was found to be a homolog of the human antiapoptotic Bcl-2 (B-cell lymphoma 2) protein. This was a groundbreaking discovery because it provides the first example that a regulator of apoptosis is conserved between worms and humans. In living cells of C. elegans, CED- 9 resides on the mitochondrial surface and tethers a CED-4 dimer, a homolog of human apoptotic protease activating factor 1 (Apaf1) that catalyzes caspase activation and apoptosis. Presumably, egl-1 transcription is repressed in living cells, and CED-3 exists as an inactive proenzyme. When a cell death specification signal induces upregulation of egl-1 transcription (see Section 3, Specification), the apoptotic cascade is initiated. EGL- 1 is a Bcl-2 homology 3 (BH3) domain-only protein that binds and induces conformational change of CED-9, causing release of CED-4 dimers. CED-4 dimers subsequently form an oligomer, which catalyzes CED-3 proenzyme autoactivation. In a particular ced-9(gf ) mutant, a glycine to glutamate substitution in the EGL-1–binding pocket of CED-9 impairs association of CED-9 with EGL- 1 and EGL-1-induced CED-4 release, thereby blocking most PCD events during C. elegans development.
(a)
(b)
10μm
Figure 34-1. Apoptotic cells (arrowheads) can be recognized by their highly refractile, raised disc morphology under di erential interference contrast (DIC) microscopy. A deficiency in engulfment of apoptotic cells caused by a ced-1 loss-of-function mutation results in increased cell corpses in embryos (B) compared with wild-type embryos (A).
Figure 34-2. The genetic pathway of programmed cell death in C. elegans. egl-1, ced-9, ced-4, and ced-3 constitute the central cell killing pathway, which culminates in the activation of the CED-3 caspase. The transcription of egl-1 is regulated by a combination of transcriptional repressors and activators that determinate life versus death fates of specific cells, such as male-specific chemosensory neurons (CEMs), hermaphrodite-specific neurons (HSNs), the P11.aaap cell, and neurosecretory motor (NSM) sister cells. Other genes such as ced-3 or ceh-30 can also serve as checkpoints for cell death specification. The autoactivation of the CED-3 proenzyme is inhibited by CSP-3. Activated CED-3 caspase coordinates various cell death execution events by cleaving and activating downstream e ectors, leading to mitochondrial elimination, DNA degradation and engulfment of apoptotic cells by phagocytes. Multiple pathways are found to mediate each of these cell death execution events.
CAENORHABDITIS ELEGANS AND APOPTOSIS |
399 |
The structure–function relationship of CED-9 is not identical to that of its mammalian counterpart. The C- terminal half of CED-9 resembles Bcl-2 and exerts its antiapoptotic activity by binding to CED-4 and preventing CED-4 from inducing CED-3 autoactivation. However, the N-terminus of CED-9, not found in Bcl-2, contains two CED-3 cleavage sites that are important for CED-9’s death inhibitory activity, probably acting as a competitive substrate for CED-3. Surprisingly, a ced-9(lf) mutation, which causes ectopic cell death, and a weak ced-3(lf) mutation, which mildly inhibits cell death, in combination result in the survival of more cells that are destined to die in the pharynx of the worm than a weak ced-3(lf) mutation alone. This suggests that ced-9 possesses a proapoptotic activity in addition to its antiapoptotic activity, though the reason for this is unclear. Currently, egl-1 and ced-9 are the only described Bcl-2 family members in C. elegans.
The primary function of CED-9 and Bcl-2 is to regulate the formation of the apoptosome, a structural scaffold of CED-4 in C. elegans and Apaf1 in mammals that activates caspase proenzymes. Though Bcl-2 does not directly bind Apaf1, it regulates the mitochondrial release of cytochrome c, a key component of the electron transport chain, which then promotes conformational change of monomeric Apaf1, binding and hydrolysis of ATP, and heptamerization into a wheel-shaped apoptosome. This results in exposure of caspase recruitment domains (CARD) in Apaf1 that engage and activate procaspase-9. Similarly, CED-4 is released from CED-9 as a dimer and forms an oligomeric apoptosome that activates the CED-3 proenzyme. This process does not require cytochrome c because CED-4 lacks the domain containing WD40 repeats that is important for Apaf1 binding to cytochrome c. Furthermore, CED-3 activation can be recapitulated in vitro using only recombinant CED-3, CED-4, CED-9, and EGL-1 in the absence of cytochrome c and adenosine triphosphate (ATP). Therefore, the involvement of cytochrome c and ATP hydrolysis in mammalian systems more tightly couples mitochondrial demise with apoptosis.
CED-3 is a member of the caspase family and likely acts by cleaving its substrates to initiate various cell death execution events. CED-3 is the only known caspase in C. elegans that has a definitive role in apoptosis, whereas multiple caspases have been implicated in apoptosis in mammalian systems. Given the essential role of ced-3 in cell killing, regulating CED- 3 proenzyme expression and activation is crucial for determining the fate of a cell. In Drosophila and mammalian systems, inhibitors of apoptosis (IAPs) negatively regulate procaspase activation and catalytic activity of
caspases. IAPs contain at least one baculoviral IAP repeat (BIR) domain that determines their caspase-binding specificity. In addition, other species contain IAP antagonists, such as Grim and Reaper in Drosophila and Smac/DIABLO in mammals. These factors bind IAPs and disrupt their activities. However, no IAP or IAP antagonist homologs have been identified in C. elegans. The first protein identified that negatively regulates the activity of CED-3 is CSP-3, which shares sequence similarity to the small subunit of CED-3. Biochemical analysis indicates that CSP-3 associates with the CED-3 proenzyme and inhibits its dimerization and autoactivation. However, CSP-3 does not block CED-3 activation induced by CED-4, nor does it inhibit the activated CED-3 protease. Therefore, it appears that C. elegans uses distinct mechanisms to regulate the activation of the CED-3 caspase and apoptosis.
Some apoptosis regulators do not function solely in apoptosis. For example, CED-4 participates in DNA damage–induced cell cycle arrest of germ cells and regulation of cell size and body size. In humans, caspase- 1, -3, and -5 are important for cytokine maturation, demonstrating another apoptosis-independent function of these proteases. Currently, there is no demonstrated role for CED-3 outside of apoptosis. Although many questions remain, the conservation of several key cell death regulators and pathways between C. elegans and mammals is apparent.
3. SPECIFICATION
In C. elegans, cell death specification initiates the killing phase in a cell-autonomous manner, generally through upregulation of egl-1 transcription. The egl-1 gene consists of a short open reading frame flanked by a promoter and a sequence downstream of the coding region that contains several important cis-regulatory elements. A specific combination of activator(s) and repressor(s) is enlisted for regulation of egl-1 transcription based on the lineage of a cell.
A unique program exists in hermaphrodite-specific neurons (HSNs) to regulate egl-1 transcription in a sex-specific manner. HSNs innervate uterine and vulval muscles to expel fertilized eggs from the vulva of hermaphrodites. In males they are unnecessary and undergo apoptosis. A unique class of egl-1(gf) mutations causes ectopic HSN death in hermaphrodites by altering residues in a TRA-1A binding site located 6.4 kilobases downstream of the egl-1 start codon. TRA-1A, a terminal sex-determination factor, is a Zinc finger transcription factor that promotes hermaphrodite development by repressing transcription of egl-1 in hermaphrodite
400 |
BRIAN L. HARRY AND DING XUE |
HSNs and male-specific genes in general. Therefore, egl- 1(gf) mutations cause derepression of egl-1 expression and ectopic HSN death in hermaphrodites (Figure 34-2). Interestingly, egl-1(gf) animals do not display cell death defects in other cell types, indicating that this TRA-1A binding site must cooperate with other cis-elements to control appropriate egl-1 expression in HSNs.
The complexity of egl-1 transcriptional regulation is also apparent in determining the fates of neurosecretory motor (NSM) neurons, two serotonergic motor neurons, and their sister cells. During embryogenesis, a precursor neuroblast divides to generate the NSM and the NSM sister cell, the latter of which undergoes apoptosis. The fates of the NSM and NSM sister cell are determined by the expression level of CES-1 (ces, cell death specification), a Snail-like Zinc finger transcriptional repressor, as well as two helix-loop-helix (HLH) DNA-binding proteins HLH-2 and HLH-3, which induce egl-1 transcription and are important for NSM sister cell death. Interestingly, the binding sites of CES-1 (Snail binding sites) and HLH-2/HLH-3 (E-box motifs) overlap, and in vitro CES-1 can compete with HLH-2/HLH- 3 for binding to these cis-elements positioned 4 kilobases downstream of the egl-1 start codon. When ces-1 is activated in NSMs, HLH-2/HLH-3 binding to egl-1 is excluded and the cells live. In NSM sister cells, ces-1 is not expressed and the HLH-2/HLH-3 complex induces cell death. The expression of ces-1 is in turn curbed by CES-2, a basic leucine zipper DNA-binding protein, and DNJ-11, a DnaJ domain-containing protein. Loss of ces-2 or dnj-11 results in transcriptional upregulation of ces-1 and ectopic survival of the NSM sister cells (Figure 34-2). Comparison of mechanisms that regulate the deaths of NSM sister cells and sex-specific HSN neurons supports a model in which egl-1 is intricately regulated by a combination of transcriptional activators and repressors.
The death of some of the ventral cord cells offers another glimpse into how egl-1 transcription is regulated in specific cells. Ventral cord precursor cells give rise to a variety of cell types, including motor neurons and hypodermal cells. One of these cells, P11.aaap (the posterior daughter of the anterior daughter of the anterior daughter of the anterior daughter of the P11 ventral cord precursor cell) normally undergoes cell death. PCD of P11.aaap requires ceh-20, which encodes the C. elegans homolog of Pbx (pre–B-cell leukemia transcription factor), and the homeobox (Hox) gene mab-5. A complex of CEH-20 and MAB-5 has been shown to bind a Pbx/Hox site in the egl-1 gene to activate egl-1 transcription. Interestingly, ceh-20 and mab-5 are also expressed in P11.aaaa, the sister of P11.aaap that lives, indicating that an additional mechanism or a transcription
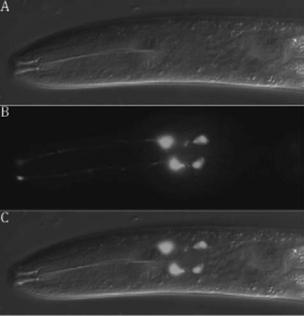
Figure 34-3. The four male-specific chemosensory (CEM) neurons located in the cephalic region of the animal undergo programmed cell death in hermaphrodites. The CEM neurons in males can be labeled by
aPpkd-2gfp reporter construct. DIC (A), GFP (B), and merged DIC/GFP
(C)images of male CEM neurons are shown. See Color Plate 44.
repressor exists to prevent egl-1 transcription in the P11.aaaa cell. Therefore, the P11.aaap cell, HSNs, and NSM sister cells use different sets of transcription factors but a similar paradigm to control egl-1 expression and activation of apoptosis.
Although the current theme of cell death specification revolves around regulation of egl-1 transcription, exceptions to this rule have been noted. C. elegans tailspike cells produce a bundle of microtubule filaments over which the cuticle of the tail forms. They are unique in that they survive more than 5 hours before they die, much longer than other cells that die during development. ced-3 and ced-4 are absolutely required for tailspike cell death, but egl-1 is only partially required. Interestingly, tail-spike cell death depends on upregulation of ced-3 by pal-1 (Figure 34-2), which encodes a homolog of the Caudal homeodomain protein that can bind the ced- 3 promoter. However, PAL-1 is expressed in many other cells that do not undergo cell death, indicating that additional factors exist either to promote tail-spike cell death or to inhibit PAL-1–induced cell death in other cells.
The death specification of male-specific chemosensory neurons (CEMs, Figure 34-3) provides yet another example of how different developmental cues can converge at a non–egl-1 checkpoint to control the life versus death fate of a cell. Surviving CEMs require the ceh-30 gene, which encodes a BarH homeodomain protein that