

4Mitochondria and Cell Death
Gavin P. McStay and Douglas R. Green
1. INTRODUCTION
Cell death pathways use genetically encoded and derived components to transduce specific death-inducing signals into a common phenotype associated with death. These signals often include steps that impinge on mitochondria as a means of initiating or amplifying the process. Because mitochondria are organelles that contain unshared lipid and protein components, this makes them ideal targets for specific integration into cell death pathways at defined steps because of the actions of proteins present in the cytosol that target and respond to these unique components. Mitochondria are the site of the cell’s major energy-generating system that produces adenosine triphosphate (ATP), used to maintain vital cellular functions. By being involved in these two conflicting processes, a cell ensures that mitochondrial energy generation and cell death are generally exclusive events. This is compatible with cellular fate ensuring shut-down of anabolic processes and favoring dismantling of cellular architecture and function, thus making death proceed in a swift manner. Here we discuss the function of mitochondria in apoptosis and necrosis and how these roles affect other aspects of mitochondrial biology.
2. MITOCHONDRIAL PHYSIOLOGY
Mitochondria are double membrane organelles that exist in all eukaryotic cells. They are termed the “powerhouse” of the cell because they provide energy in the form of ATP to ensure that essential cellular processes are maintained and cells can survive and/or proliferate in their surroundings. ATP generation occurs through the electron transport chain in the inner mitochondrial membrane (IMM). Metabolites derived from the tricarboxylic acid (TCA) cycle undergo a series of
enzymatic reactions generating reduced nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide (FADH2). Electrons from NADH are transported from complex I (NADH:quinone oxidoreductase) to coenzyme Q, to complex III (coenzyme Q:cytochrome c oxidoreductase), then finally cytochrome c to complex IV (cytochrome c oxidase), which uses these electrons to generate water from molecular oxygen. FADH2 is generated by complex II (succinate:coenzyme Q oxidoreductase), and electrons are also transferred to coenzyme Q and follow the same pathway as NADH-derived electrons. Complexes I, III, and IV pump protons from the matrix into the intermembrane space (IMS), creating a pH and electrical gradient across the IMM (positive and acidic in the IMS, negative and basic in the matrix). This gradient is termed the mitochondrial membrane potential and is coupled to the function of many mitochondrial processes, such as protein, ion, or metabolite transport across the IMM. Regarding the generation of ATP, the IMM enzyme F1F0 ATP synthase drives ATP synthesis against the flow of protons from the IMS to the matrix. The IMM has a larger surface area and is folded many times within the outer mitochondrial membrane (OMM). This folding creates sub-organellar compartments known as cristae that are enriched in components of the electron transport chain, particularly cytochrome c. Mitochondria are the main source of reactive oxygen species (ROS) in a cell, mostly through the inefficient transfer of electrons between components of the electron transport chain. Therefore, mitochondria are very susceptible to the damaging effects of these highly reactive intermediates. Damage to mitochondrial proteins, lipids, and/or DNA must be efficiently repaired to maintain proper function. However, mitochondria that have received too much damage may be removed entirely through mitochondrial-specific autophagy (mitophagy)
37
38 |
GAVIN P. MC STAY AND DOUGLAS R. GREEN |
or can fuse with less damaged counterparts, allowing damaged proteins, lipids, or DNA to be diffused or repaired. Mitochondrial fusion and fission events allow the mitochondrial network to remain a relatively homogeneous population and ensure equal division of mitochondria during cell division.
3. THE MITOCHONDRIAL PATHWAY OF APOPTOSIS
Several cell death pathways impinge on mitochondria as a means of initiating or amplifying cell death signaling. During the decision processes of apoptotic cell death, the BCL-2 family of proteins is engaged to regulate the release of mitochondrial IMS proteins, such as cytochrome c, into the cytosol in a process termed mitochondrial outer membrane permeabilization (MOMP). MOMP engages the mitochondrial pathway of apoptosis mainly by the release of cytochrome c. On its release, cytochrome c induces the activation of caspases, a family of proteases that cleave hundreds of intracellular substrates. This activity ensures that dying cells targeted by apoptosis are dismantled, recognized and removed in a manner that minimizes damage to the surrounding tissue.
4. MITOCHONDRIAL OUTER MEMBRANE
PERMEABILIZATION
Mitochondria are engaged in the apoptotic pathway through the action of the BCL-2 family (described in detail elsewhere in this volume) characterized by one or more BCL-2 homology domains (BH1–4). Some proapoptotic members of this family contain only the third BH domain and are known as the BH3-only proteins; specific members of this subset include BID, BIM, PUMA, and BAD, among others. The other type of proapoptotic proteins are known as the multidomain proapoptotic effector proteins. This subset is composed of the pore-forming proteins BAX and/or BAK. The antiapoptotic BCL-2 proteins contain all four BH domains and include the proteins BCL-2, BCL-xL, and MCL- 1. A subset of BH3-only proteins can directly activate BAX or BAK inducing pore formation in the OMM (Figure 4-1). Antiapoptotic proteins inhibit the activity of BH3-only proteins by binding and thus preventing activation of BAX and BAK. Other members of the BH3only proteins can only bind to the antiapoptotic proteins and are able to displace bound direct activators that can activate BAX and BAK. Once activated, BAX and BAK are thought to form oligomeric pores that allow the release of IMS proteins, including cytochrome c, into the cytosol. The OMM acts as a platform for BAX and BAK
activation by the presence of specific components. This is demonstrated as lipid vesicles require the mitochondrial lipid, cardiolipin, or resident OMM proteins to be permeabilized by BAX, and vesicles lacking cardiolipin or resident OMM proteins resist permeabilization.
5. MORPHOLOGICAL CHANGES IN MITOCHONDRIA
DURING MOMP
A frequent feature of the mitochondrial network during apoptosis is that it undergoes fragmentation, changing from a reticular network to punctate structures in the cell (Figure 4-2). The importance of this event has not been elucidated but has led to the hypothesis that proteins involved in mitochondrial dynamics participate in BAX/BAK activation and MOMP. Dynaminrelated protein-1 (DRP-1) is responsible for constricting the OMM and IMM to induce mitochondrial fission. Removing DRP-1 function can inhibit or delay MOMP and apoptosis. However, the mechanism of inhibition may not be due to inhibition of mitochondrial fission but rather directly on the BCL-2 family. Evidence suggests that fused mitochondria are able to release cytochrome c after apoptotic stimuli, supporting the role of the mitochondrial fission and fusion proteins as regulators of BAX/BAK activation and not fission or fusion per se as a mediator of MOMP.
Another protein involved in mitochondrial dynamics implicated in MOMP regulation is optic atrophy-1 (OPA- 1). This protein resides in the IMM and IMS as either short or long isoforms derived from differential splicing and proteolysis. OPA-1 is involved in both the fusion of IMMs and organization of cristae. Cristae contain approximately 85% of total cytochrome c in mitochondria, whereas approximately 15% is localized to the IMS directly beneath the OMM. OPA-1 exists as oligomers (possibly as a trimer made up of two IMM-bound isoforms and one IMS-soluble isoform) at the cristae openings (Figure 4-1). On BAX or BAK activation, these complexes dissociate, allowing the efflux of cytochrome c from the cristal space that is now continuous with the IMS and eventually through the BAX/BAK pore at the OMM. Inhibition of OPA-1 expression supports enhanced cytochrome c release and apoptosis, whereas OPA-1 overexpression is suggested to inhibit MOMP.
6. DOWNSTREAM OF MITOCHONDRIAL OUTER
MEMBRANE PERMEABILIZATION
The actions of BAX or BAK on the OMM cause the release of many proteins from the IMS (Figure 4-2). Once MOMP has occurred, cytochrome c, which normally
MITOCHONDRIA AND CELL DEATH |
39 |
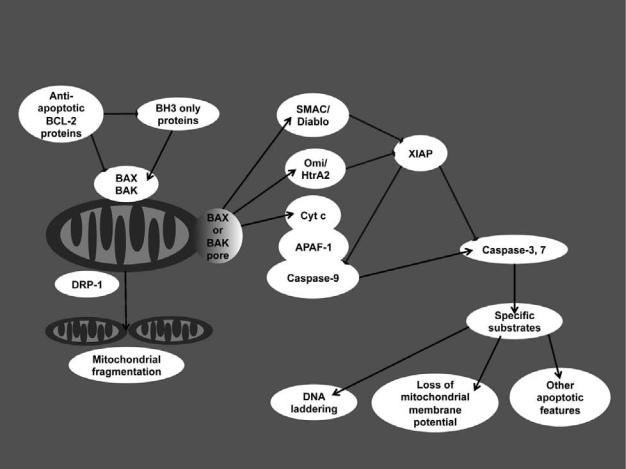
Figure 4-1. Events that occur at the inner and outer mitochondrial membranes upstream and downstream of mitochondrial outer membrane permeabilization. C9, caspase-9; C3/7, caspase-3/caspase-7; cyt c, cytochrome c; ETC, electron transport chain; IMM, inner mitochondrial membrane; IMS, intermembrane space; MOMP, mitochondrial outer membrane permeabilization; OMM, outer mitochondrial membrane. See text for full description.
resides in the IMS, is released into the cytosol, resulting in the activation of caspases. In the cytosol, cytochrome c binds to apoptotic protease activating factor-1 (APAF- 1), which in concert with dATP causes a conformational change in APAF-1, which results in oligomerization into a heptameric structure called the apoptosome (Figure 4-2). The apoptosome acts as a scaffold for the recruitment and activation of procaspase-9. Dimerization of caspase-9, via the apoptosome, induces its proteolytic activity. Caspase-9 then cleaves and thereby activates caspase-3 and -7, proteases responsible for the cleavage of many cellular proteins that result in the phenotypic hallmarks of apoptosis, such as cutting of DNA into small fragments, condensation of chromatin in the nucleus, dissipation of mitochondrial membrane potential, and redistribution of phosphatidylserine (PS) from the inner leaflet to the outer leaflet of the plasma membrane.
Other proteins accompany cytochrome c during MOMP. These include SMAC/Diablo (second mitochondrial activator of apoptosis/direct IAP binding protein with low pI) and Omi/HtrA2, both of which assist in caspase activation by antagonizing the inhibitor of apoptosis proteins (IAPs), a family of proteins that inhibit caspases directly. Activated caspases-3, -7 and -9 are potently inhibited by X-linked inhibitor of apoptosis protein (XIAP), but this inhibition can be relieved by the action of IAP antagonists, especially SMAC/Diablo through its IAP-binding motif (IBM) that disrupts IAP:caspase complexes. Therefore, the main role of SMAC/Diablo may be ensuring a concerted burst of caspase activity downstream of MOMP. Omi/HtrA2 also contains an IBM and may inhibit IAP function through a similar mechanism.
There are two other proteins that may play important roles in cell death on release from mitochondria;
40 |
GAVIN P. MC STAY AND DOUGLAS R. GREEN |
Figure 4-2. Events that occur downstream of mitochondrial outer membrane permeabilization with focus on proteins released from the intermembrane space and their e ects on cytosolic components. cyt c, cytochrome c; XIAP, X-linked inhibitor of apoptosis protein. See text for full description.
however, their mechanisms of release are unclear. Apoptosis-inducing factor (AIF) exists in the IMM and appears to play a role in mitochondrial complex I assembly or function. Once MOMP has occurred, AIF can translocate from mitochondria into the cytosol, but this event may depend on caspases and/or calpains, placing AIF as a mediator of cellular dismantling rather than as an initiator of cell death. AIF has also been suggested to act as a direct mediator of DNA fragmentation. Lastly, endonuclease G, which is present in the mitochondrial matrix, is reportedly released after MOMP, and this protein might also cause DNA fragmentation independently of caspases. Other proteins have been described that are released during MOMP (e.g., adenylate kinase-2), but these may be only bystanders with no specific proapoptotic function.
Disruption of the OMM dilutes cytochrome c, but sufficient protein can be present to sustain electron transport. However, activated caspases gain access to the IMS and can specifically disrupt mitochondrial function.
Oxidative phosphorylation activated by complexes I and II of the electron transport chain are inhibited when caspases are activated. Caspase-3 cleaves and inactivates complex I at a specific site in the subunit NDUFS1 (NADH dehydrogenase [ubiquinone[ Fe [Iron]–sulfur 1) (Figure 4-1). This inhibits complex I activity and disrupts electron transport and ultimately mitochondrial function through dissipation of the mitochondrial membrane potential. Preventing the cleavage of NDUFS1 subunit delays apoptosis and delays PS exposure on the plasma membrane, perhaps through maintaining ATP levels in the cell. Also, mitochondria that have undergone MOMP are targets for mitophagy. This removes the damaged organelles from cells and its detrimental effects such as ROS generation or excessive substrate (such as ATP) consumption.
Even though MOMP usually leads to caspase activation, most cells die, even if caspase activity is defective. In situations in which caspase inhibitors are present or caspase-activating proteins (e.g., APAF-1) are absent,
MITOCHONDRIA AND CELL DEATH |
41 |
cells submit to caspase-independent cell death (CICD). Cells die in this process as a result of the loss of energyproducing capability of mitochondria as the mitochondrial membrane potential slowly dissipates because of impaired oxidative phosphorylation. To maintain mitochondrial membrane potential, ATP is hydrolyzed by F1F0 ATP synthase, but eventually ATP is depleted, and this leads to a slow, uncontrolled, necrotic death. This process can be inhibited by over-expression of glyceraldehyde 3-phosphate dehydrogenase (GAPDH), a glycolytic enzyme that is able to sustain ATP concentrations through glycolysis and also engage autophagy to remove mitochondria that have undergone MOMP. Cells undergoing CICD are able to proliferate after MOMP when caspase activation is inhibited and GAPDH is overexpressed, suggesting that sustaining energy production and removal of damaged mitochondria is enough to ensure cell survival after MOMP.
7. MITOCHONDRIAL PERMEABILITY TRANSITION PORE
AND NECROTIC CELL DEATH
As mitochondria are responsible for energy production they are able to sense the metabolic status of a cell. In pathological situations such as ischemia-reperfusion, when blood flow is temporarily restricted followed by return to normal blood flow, or other prolonged cellular stresses when metabolic substrates become limiting or metabolic end-products accumulate, mitochondria respond by opening the mitochondrial permeability transition pore (MPTP). This pore forms in the IMM and is composed, in part, of cyclophilin D (CyP-D) in the matrix. Other suggested components include the adenine nucleotide translocase (ANT), but this is controversial (Figure 4-3). The opening of the MPTP allows exchange of mitochondrial matrix and cytosolic components of less than 1,500 D, destroying the compartmentalization of the mitochondria, as evidenced by the dissipation of the mitochondrial membrane potential. Loss of the mitochondrial membrane potential prevents most functions of mitochondria. In the absence of a membrane potential, mitochondria in a cell are no longer able to produce ATP using oxidative phosphorylation and will eventually die through necrosis when critical functions are lost because of the decrease of ATP levels.
The composition of the MPTP is still unresolved, but many candidates have been proposed. The most convincing component of the MPTP is the matrix peptidylprolyl cis-trans isomerase cyclophilin D (CyP-D). In mice that lack this protein, MPTP formation is virtually undetectable, and inhibitors of this enzyme prevent MPTP. Cells derived from CyP-D–deficient mice show
inhibition of death in situations of necrosis induced by hydrogen peroxide or ischemia reperfusion in isolated hearts. However, cells from these mice do not show any resistance to apoptotic stimuli, demonstrating that the MPTP is not involved in releasing cytochrome c during apoptosis.
CyP-D is not thought to be the pore-forming unit of the MPTP, but rather an initiator of a conformational change in an IMM protein. Most speculation has focused on the adenine nucleotide translocase (ANT), the mediator of ADP/ATP exchange between the mitochondrial matrix and cytoplasm. This is mainly due to the number of known ligands of the ANT that influence MTPTP opening. However, mitochondria from mice lacking all isoforms of this protein can still undergo MPTP opening, leading to the suggestion that another component may be the pore-forming unit (e.g., the phosphate carrier). Alternatively, it is possible that many similar proteins may be able to form the MPTP, especially members of the mitochondrial carrier family, of which the preceding two candidates are members. This has also led to the hypothesis that the pore is not formed by a specific protein but is due to misfolding of IMM proteins that are induced by the stresses known to engage the MPTP. In addition, OMM components may be able to influence the opening of the MPTP, such as VDAC, the peripheral benzodiazepine receptor, and creatine kinase, mainly based on experiments using specific ligands of these components or through the isolation of proteins complexes that contain putative components of the MPTP. Their roles in the MPTP remain controversial.
The MPTP forms in the IMM under certain conditions mainly driven by accumulation of excess calcium ions in the mitochondrial matrix and can be sensitized by metabolic changes within mitochondria, such as oxidative stress, adenine nucleotide depletion, high mitochondrial matrix pH, or low mitochondrial membrane potential. Insults such as ischemia-reperfusion in the heart or brain result in a large wave of necrotic cell death that is initiated by metabolic stress within the affected organ, such as bursts of reactive oxygen species and depletion of metabolites, known activators of MPTP opening.
8. COMPARISON OF THE VERTEBRATE AND INVERTEBRATE
PATHWAYS OF MITOCHONDRIAL CELL DEATH
The mitochondrial pathway of apoptosis, described previously, is prevalent in vertebrate organisms; however, other pathways with homologous proteins exist in invertebrates. Differences exist between two model organisms, the nematode (Caenorhabditis elegans) and the

42 |
GAVIN P. MC STAY AND DOUGLAS R. GREEN |
Figure 4-3. Mitochondrial changes associated with mitochondrial permeability transition pore opening. ADP/ATP, adenosine di/tri-phosphate; ANT, adenine nucleotide translocase; Ca2+ trans., Ca2+ uniporter; CyP- D, cyclophilin-D; ETC, electron transport chain; F1F0, F1F0 ATP synthase; IMM, inner mitochondrial membrane; IMS, intermembrane space; MPTP, mitochondrial permeability transition pore; OMM, outer mitochondrial membrane; VDAC, voltage-dependent anion channel. See text for full description.
fruit fly (Drosophila melanogaster). In C. elegans, apoptosis occurs by a pathway that does not rely on MOMP, but mitochondria appear to play a role. In this pathway, the antiapoptotic BCL-2 homolog cell death abnormality (CED)-9 exists in a complex with CED-4 (the APAF-1 homolog) at the OMM. This complex is disrupted by the BH3-only homolog EGL-1, allowing activation of CED-4 and ensuing activation of the caspase-3 homolog CED- 3. CED-9 has also been shown to regulate mitochondrial fission and fusion, similar to the effects of the mammalian BCL-2 family on mitochondria. Another similarity to mammalian apoptosis is the fragmentation of mitochondria during apoptosis in C. elegans. This is due to EGL-1–mediated binding to CED-9 and regulation of the C. elegans DRP-1 or mitofusin-2 homologs. Moreover, enhanced expression of DRP-1 has been suggested to induce mitochondrial fragmentation and cell death. Ectopic expression of CED-9 in mammalian cells
is able to induce mitochondrial fusion, but is unable to inhibit release of IMS proteins after induction of apoptosis, indicating that regulation of mitochondrial fission and fusion by the BCL-2 family is conserved between C. elegans and vertebrates.
The same holds true for apoptosis in D. melanogaster, where mitochondrial fragmentation has been observed. As in C. elegans, release of proteins from the IMS in D. melanogaster is not an essential process that is required for apoptosis. Although cytochrome c release has been observed during apoptosis in certain cell types in drosophila, this is a caspase-dependent event, indicating that it is a consequence of apoptosis and not an inducing event. However, the Drosophila homolog of DRP-1 has been shown to be involved in mitochondrial fragmentation during apoptosis. Similar to C. elegans, mitochondria in Drosophila act as a platform for apoptosis activation events as the proapoptotic IAP