

IMPLICATIONS OF NITROSATIVE STRESS-INDUCED PROTEIN MISFOLDING IN NEURODEGENERATION |
149 |
by which NO induces protein misfolding and ER stress, however, have remained enigmatic until recently.
Interestingly, we have recently reported that excessive NO can also lead to S-nitrosylation of the activesite thiol groups of PDI, and this reaction inhibits both its isomerase and chaperone activities (Figure 13-2). Mitochondrial complex I insult by rotenone can also result in S-nitrosylation of PDI in cell culture models. Moreover, we found that PDI is S-nitrosylated in the brains of virtually all cases examined of sporadic AD and PD. Under pathological conditions, it is possible that both cysteine sulfhydryl groups in the active sites of PDI form S-nitrosothiols. Additionally, we speculate that vicinal (nearby) cysteine thiols reacting with NO can form nitroxyl disulfide, and such reaction may potentially occur in the catalytic side of PDI to inhibit enzymatic activity. To determine the consequences of S-nitrosylated PDI (SNO-PDI) formation in neurons, we exposed cultured cerebrocortical neurons to neurotoxic concentrations of NMDA, thus inducing excessive Ca2+ influx and consequent NO production from nNOS. Under these conditions, we found that PDI was S-nitrosylated in a NOS-dependent manner. SNOPDI formation led to the accumulation of polyubiquitinated/misfolded proteins and activation of the UPR. Moreover, S-nitrosylation abrogated the inhibitory effect of PDI on aggregation of proteins observed in Lewy body inclusions. S-Nitrosylation of PDI also prevented its attenuation of neuronal cell death triggered by ER stress, misfolded proteins, or proteasome inhibition (Figure 13-2).
The UPS and UPR are apparently impaired in the aging brain. Additionally, inclusion bodies similar to those found in neurodegenerative disorders can appear in brains of normal aged individuals or those with subclinical manifestations of disease. These findings suggest that the activity of the UPS and molecular chaperones may decline in an age-dependent manner. Given that we have not found detectable quantities of SNO-parkin and SNO-PDI in normal aged brain, we speculate that S-nitrosylation of these and similar proteins may represent a key event that contributes to susceptibility of the aging brain to neurodegenerative conditions.
7. POTENTIAL TREATMENT OF EXCESSIVE NMDA-INDUCED CA2+ INFLUX AND FREE RADICAL GENERATION
One mechanism that could potentially curtail excessive Ca2+ influx and resultant overstimulation of nNOS activity, with resultant S-nitrosylation of parkin, PDI, and other proteins, would be inhibition of NMDA receptors. Until recently, however, drugs in this class blocked
virtually all NMDA receptor activity, including physiological activity, and therefore manifest unacceptable side effects by inhibiting normal functions of the receptor. For this reason, many previous NMDA receptor antagonists have disappointingly failed in advanced clinical trials conducted for a number of neurodegenerative disorders. In contrast, studies in our laboratory first showed that the adamantane derivative, memantine, preferentially blocks excessive (pathological) NMDA receptor activity while relatively sparing normal (physiologic) activity. Memantine does this in a surprising fashion because of its low (micromolar) affinity, even though its actions are quite selective for the NMDA receptor at that concentration (Figure 13-3). “Apparent” affinity of a drug is determined by the ratio of its “onrate” to its “off-rate” for the target. The on-rate is not only a property of drug diffusion and interaction with the target, but also the drug’s concentration. In contrast, the off-rate is an intrinsic property of the drugreceptor complex, unaffected by drug concentration. A relatively fast off-rate is a major contributor to memantine’s low affinity for the NMDA receptor. The inhibitory activity of memantine involves blockade of the NMDA receptor-associated ion channel when it is excessively open (termed open-channel block). The unique and subtle difference of the memantine blocking sites in the channel pore may explain the advantageous properties of memantine action.
Also critical for the clinical tolerability of memantine is its uncompetitive mechanism of action. An uncompetitive antagonist can be distinguished from a noncompetitive antagonist, which acts allosterically at a noncompetitive site (i.e., at a site other than the agonistbinding site). An uncompetitive antagonist is defined as an inhibitor whose action is contingent on prior activation of the receptor by the agonist. Hence the same amount of antagonist blocks higher concentrations of agonist relatively better than lower concentrations of agonist. Some open-channel blockers function as pure uncompetitive antagonists, depending on their exact properties of interaction with the ion channel. This uncompetitive mechanism of action coupled with a relatively fast off-rate from the channel yields a drug that preferentially blocks NMDA receptor-operated channels when they are excessively open while relatively sparing normal neurotransmission. In fact, the relatively fast off-rate is a major contributor to a drug like memantine’s low affinity for the channel pore. Although many factors determine a drug’s clinical efficacy and tolerability, it appears that the relatively rapid off-rate is a predominant factor in memantine’s tolerability in contrast to other NMDA-type receptor antagonists. Thus the
150 |
TOMOHIRO NAKAMURA AND STUART A. LIPTON |
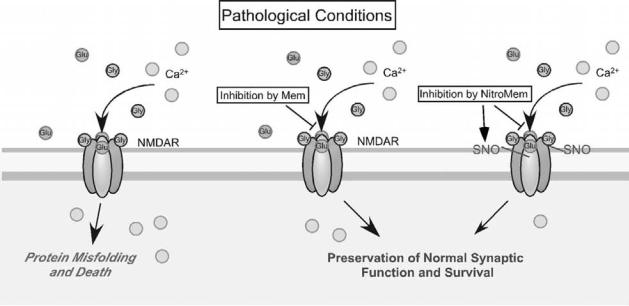
Figure 13-3. Memantine and NitroMemantine preferentially block excessive extrasynatpic NMDA receptor activity. (Left) Excessive activation of the NMDA receptor (NMDAR), predominantly at extrasynaptic sites, is thought to induce neuronal cell injury and death and is associated with the accumulation of misfolded proteins. (Middle and Right) Memantine (Mem) and the newer NitroMemantines (NitroMem) preferentially block excessive (pathological extrasynaptic) NMDA receptor activity while relatively sparing normal (physiologic synaptic) activity. (Right) NitroMemantines target an NO species to the NMDA receptor by tethering a nitro group to the memantine moiety and thus add to the neuroprotective action of memantine by S-nitrosylating the receptor.
critical features of memantine’s mode of action are its Uncompetitive mechanism and Fast Off-rate, or what we call a UFO drug – a drug that is present at its site of inhibitory action only when you need it and then quickly disappears. Memantine has been used for many years in Europe to treat PD, and regulatory agencies in both Europe and the United States recently voted its approval as the first treatment for moderate-to-severe AD. It is currently under study for a number of other neurodegenerative disorders.
As promising as the results with memantine are, we are continuing to pursue ways to use additional modulatory sites on the NMDA receptor to block excitotoxicity even more effectively and safely than memantine alone. New approaches in this regard are explored next.
8. FUTURE THERAPEUTICS: NITROMEMANTINES
NitroMemantines are second-generation memantine derivatives that are designed to have enhanced neuroprotective efficacy without sacrificing clinical tolerability. As mentioned earlier, a nitrosylation site(s) is located on the extracellular domain of the NMDA receptor, and S-nitrosylation of this site (i.e., NO reaction with the sulfhydryl group of a critical cysteine residue) downregulates (but does not completely shut off) receptor
activity (Figure 13-3). The drug nitroglycerin, which generates NO-related species, can act at this site to limit excessive NMDA receptor activity. In fact, in rodent models, nitroglycerin can limit ischemic damage , and there is some evidence that patients taking nitroglycerin for other medical reasons may be resistant to glaucomatous visual field loss. Consequently, we carefully characterized the S-nitrosylation sites on the NMDA receptor to determine whether we could design a nitroglycerinlike drug that could be more specifically targeted to the receptor. In brief, we found that five different cysteine residues on the NMDA receptor could interact with NO. One of these, located at cysteine residue 399 (Cys399) on the NR2A subunit of the NMDA receptor, mediates ≥90% of the effect of NO under our experimental conditions. From crystal structure models and electrophysiologic experiments, we further found that NO binding to the NMDA receptor at Cys399 may induce a conformational change in the receptor protein that makes glutamate and Zn2+ bind more tightly to the receptor. The enhanced binding of glutamate and Zn2+ in turn causes the receptor to desensitize and, consequently, the ion channel to close. Electrophysiologic studies have demonstrated this inhibitory effect of NO on the NMDA receptor-associated channel. Moreover, as the oxygen tension is lowered (a pO2 of 10–20 torr is found in
IMPLICATIONS OF NITROSATIVE STRESS-INDUCED PROTEIN MISFOLDING IN NEURODEGENERATION |
151 |
normal brain, and even lower levels under hypoxic/ ischemic conditions), the NMDA receptor becomes more sensitive to inhibition by S-nitrosylation.
Unfortunately, nitroglycerin itself is not very attractive as a neuroprotective agent. The same cardiovascular vasodilator effect that makes it useful in the treatment of angina could cause dangerously large drops in blood pressure in patients with dementia, stroke, traumatic injury, or glaucoma. However, the open-channel block mechanism of memantine not only leads to a higher degree of channel blockade in the presence of excessive levels of glutamate, but also can be used as a homing signal for targeting drugs (e.g., the NO group) to hyper-activated, open NMDA-gated channels. We have therefore been developing combinatorial drugs (NitroMemantines) that theoretically should be able to use memantine to target NO to the nitrosylation sites of the NMDAR to avoid the systemic side effects of NO. Two sites of modulation would be analogous to having two volume controls on your television set for fine-tuning the audio signal.
Preliminary studies have shown NitroMemantines to be highly neuroprotective in both in vitro and in vivo animal models. In fact, they appear to be more effective than memantine at lower dosage. Moreover, because of the targeting effect of the memantine moiety, NitroMemantines appear to lack the blood pressure–lowering effects typical of nitroglycerin. More research still needs to be performed on NitroMemantine drugs, but by combining two clinically tolerated drugs (memantine and nitroglycerin), we have created a new, improved class of UFO drugs that should be both clinically tolerated and neuroprotective.
9. CONCLUSIONS
Excessive nitrosative and oxidative stress triggered by excessive NMDA receptor activation and/or mitochondrial dysfunction may result in malfunction of the UPS or molecular chaperones, thus contributing to abnormal protein accumulation and neuronal damage in sporadic forms of neurodegenerative diseases. Our elucidation of an NO-mediated pathway to dysfunction of parkin and PDI by S-nitrosylation provides a mechanistic link between free radical production, abnormal protein accumulation, and neuronal cell injury in neurodegenerative disorders such as PD. Elucidation of this new pathway may lead to the development of additional new therapeutic approaches to prevent aberrant protein misfolding by targeted disruption or prevention of nitrosylation of specific proteins such as parkin and PDI. This article also describes the action of memantine
via uncompetitive antagonism of the NMDA receptor with a fast off-rate. NitroMemantines enhance the neuroprotective efficacy over memantine at a given dose owing to its additional ability to S-nitrosylate the NMDA receptor. These drugs preferentially inhibit pathologically activated NMDA receptor while preserving its normal synaptic function; thus they are clinically tolerated. In this chapter we propose that the next generation of CNS drugs will interact with their target only during states of pathological activation and not interfere with the target if it is functioning properly. In the future, such perspectives should lead to additional novel, clinically tolerated neuroprotective therapeutics.
ACKNOWLEDGMENTS
This work was supported in part by a JSPS Postdoctoral Fellowship for Research Abroad (to T.N.); National Institutes of Health grants P01 HD29587, R01 EY05477, and R01 EY09024; the American Parkinson’s Disease Association, San Diego Chapter; and an Ellison Senior Scholars Award in Aging (to S.A.L.).
SUGGESTED READINGS
Ankarcrona, M., Dypbukt, J. M., Bonfoco, E., et al. (1995). Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron, 15, 961–73.
Bonfoco, E., Krainc, D., Ankarcrona, M., et al. (1995). Apoptosis and necrosis: two distinct events induced, respectively, by mild and intense insults with N-methyl-D-aspartate or nitric oxide/superoxide in cortical cell cultures. Proc Natl Acad Sci U S A, 92, 7162–6.
Chen, H. S. and Lipton, S. A. (2006). The chemical biology of clinically tolerated NMDA receptor antagonists. J Neurochem, 97, 1611–26.
Chung, K. K., Thomas, B., Li, X., et al. (2004). S-nitrosylation of parkin regulates ubiquitination and compromises parkin’s protective function. Science, 304, 1328–31.
Dawson, V. L., Dawson, T. M., London, E. D., et al. (1991). Nitric oxide mediates glutamate neurotoxicity in primary cortical cultures. Proc Natl Acad Sci U S A, 88, 6368–71.
Ellgaard, L. and Ruddock, L. W. (2005). The human protein disulphide isomerase family: substrate interactions and functional properties. EMBO Rep, 6, 28–32.
Gruber, C. W., Cemazar, M., Heras, B., et al. (2006). Protein disulfide isomerase: the structure of oxidative folding. Trends Biochem Sci, 31, 455–64.
Hess, D. T., Matsumoto, A., Kim, S. O., et al. (2005). Protein S- nitrosylation: purview and parameters. Nat Rev Mol Cell Biol, 6, 150–66.
Lipton, S. A. (2006). Paradigm shift in neuroprotection by NMDA receptor blockade: memantine and beyond. Nat Rev Drug Discov, 5, 160–70.
152 |
TOMOHIRO NAKAMURA AND STUART A. LIPTON |
Lipton, S. A., Choi, Y. B., Pan, Z. H., et al. (1993). A redoxbased mechanism for the neuroprotective and neurodestructive effects of nitric oxide and related nitroso-compounds.
Nature, 364, 626–32.
Lipton, S. A. and Rosenberg, P. A. (1994). Excitatory amino acids as a final common pathway for neurologic disorders. N Engl J Med, 330, 613–22.
Ross, C. A. and Pickart, C. M. (2004). The ubiquitin-proteasome pathway in Parkinson’s disease and other neurodegenerative diseases. Trends Cell Biol, 14, 703–11.
Shimura, H., Hattori, N., Kubo, S., et al. (2000). Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet, 25, 302–5.
Uehara, T., Nakamura, T., Yao, D., et al. (2006). S-nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature, 441, 513–7.
Yao, D., Gu, Z., Nakamura, T., et al. (2004). Nitrosative stress linked to sporadic Parkinson’s disease: S-nitrosylation of parkin regulates its E3 ubiquitin ligase activity. Proc Natl Acad Sci U S A, 101, 10810–4.
14 Mitochondrial Mechanisms of Neural Cell Death
in Cerebral Ischemia
Lucian Soane, Brian M. Polster, and Gary Fiskum
1. CELL DEATH AFTER CEREBRAL ISCHEMIA
AND REPERFUSION
Millions die annually from ischemic brain damage caused by stroke, subarachnoid hemorrhage, head trauma, shock, and postischemic injury after resuscitation from cardiac arrest. Many thousands of others either die or suffer permanent neurological impairment after surgical procedures that carry a risk for inducing cerebral hypoxia/ischemia. Neurological morbidity and mortality are primarily the consequences of necrotic, apoptotic, and other forms of neuronal and nonneuronal cell death. The most effective neuroprotective interventions must therefore target mechanisms that contribute to each of these pathways.
Traditionally, brain cell death after cerebral ischemia has been considered to be primarily necrotic in nature. More recent research revealed, however, that the pattern of cell death in the postischemic brain is much more complex. On the basis of morphology alone, both necrotic and apoptotic death mechanisms and (according to recent studies) autophagy seem to contribute to some extent to the demise of injured cells. The extent of contribution of each of these mechanisms depends on the intensity and type of injury, whether it is due to focal or transient global cerebral ischemia, the brain region(s) affected, time post-injury, and so forth. Although necrosis predominates in the center of a focal ischemic lesion (ischemic core) and can occur early (within minutes), cells with apoptotic morphology are generally reported in the penumbral region and peak at later stages (days, even weeks). In contrast, initiation of apoptotic molecular pathways has been observed within 30 minutes of reperfusion after global cerebral ischemia caused by cardiac arrest, even though most neurons that are dead or dying 24 hours later appear
necrotic. A similar course of events has been described for the core region of ischemic strokes. It has been noted, however, that in the postischemic brain, dying neurons often assume morphologies that do not reflect purely apoptotic or necrotic characteristic features, but incomplete (apoptoticor necrotic-like) or mixed phenotypes. Accordingly, some studies have proposed the existence of a necrosis-apoptosis continuum. Multiple cell death mechanisms can be activated in the injured cells, and in some models even within the same cells. The phenotype acquired by the dying cells reflects thus the degree of completion of these death pathways (i.e., availability of adenosine triphosphate [ATP] is required for completion of apoptosis). It has been suggested that a process of secondary necrosis results from rapid failure to fully develop the apoptotic pathways due to rapid depletion of energy stores. Such shifts from apoptosis to necrosis have also been demonstrated in in vitro models, and many studies begin to reveal the existence of extensive cross-communication between various cell death pathways. Although necrosis has been considered for a long time just as an accidental, unregulated form of cell death, there is now substantial evidence indicating that similar to other death pathways (i.e., apoptosis), execution of at least certain forms of necrosis is also highly regulated. Several specific mediators of necrotic death have been characterized, including RIP1 kinase induced by death receptor (DR) ligands, poly-(ADP-ribose) polymerase (PARP) 1 overactivation, and multiple mitochondrial alterations (i.e., mitochondrial permeability transition [MPT], reactive oxygen species [ROS] production; see next section) (Figure 14-1). The term necroptosis has been proposed to distinguish the regulated necrotic death from accidental necrosis. A central role in coordinating many of the cell death pathways implicated in ischemia/reperfusion is held by mitochondria.
153