

30 |
HENNING WALCZAK AND CHAHRAZADE KANTARI |
for the best possible clinical use of the different TRAIL receptor agonists, it is of pivotal importance that we spend more time and effort on the thorough understanding of the biochemical mechanisms of TRAIL apoptosis resistance versus sensitivity of different types of cancer cells that rely on specific combinations of alterations in the expression of a particular set of oncogenes and tumor suppressor genes as compared with normal cells. Thereby we may be able in the future to specifically target expression or activity of certain factors to achieve cancer-specific TRAIL apoptosis sensitization on the level of the individual cancer patient. Novel proteomic and genomic technologies and the integration of the results obtained by their application in intelligent systems biology approaches will most likely be instrumental in uncovering the mechanisms that govern TRAIL apoptosis sensitivity versus resistance in different types of cancer. These studies will enable a more targeted and individualized use of TRAIL receptor agonists and combination with other drugs in cancer therapy in the future.
3. DEATH RECEPTOR–LIGAND SYSTEMS WITH PRIMARILY
IMMUNOSTIMULATORY, PROINFLAMMATORY ACTIVITY
3.1. The TNF system
3.1.1. Biochemistry of TNF signal transduction
The founding member of the TNFSF is a homotrimer of TNF molecules, each 157 amino acids in length. The trimer adopts a characteristic conformation, which is now commonly referred to as the TNF fold. TNF is mainly produced by activated macrophages. Depending on the physiologic or pathological context, it is, however, also expressed by a number of other cell types. The binding of TNF to its receptors triggers a series of intracellular events that primarily induces the activation of NF-κB and the mitogen-activated protein (MAP) kinases c-Jun N-terminal kinase (JNK) and p38. These events lead to immunostimulatory gene induction, which often drives an inflammatory response (Figure 3-3). Induction of apoptosis by TNF is only a secondary signal (see Section 5).
The diverse biological effects of TNF are mediated by two different receptors, TNF-R1 and TNF-R2. Although TNF-R1 is expressed on cells of almost all tissues, TNFR2 is almost exclusively present on cells of lymphoid origin. TNF-R1 contains a DD and initiates the majority of TNF-induced biological activities, including induction of cell death by apoptosis. Yet TNF-R2 was also shown to be capable of inducing apoptosis. It has now been demonstrated, however, that TNF-R2–induced apopto-
sis works via an indirect loop mechanism: TNF-R2 crosslinking induces expression of TNF, which then binds to TNF-R1 to induce cell death. Apart from inducing TNF, the TNF-R2–mediated signal also sensitizes cells to TNF-R1–mediated apoptosis by depleting TRAF2 and cIAPs, which causes the gene-inducing capacity of TNFR1 to be diminished, strengthening the apoptotic arm of the response (Figure 3-4). Thus the TNF-R2 signal is a modulator of the TNF-R1 signal transduction machinery, and other non–DD-containing receptors of the TNFRSF described to induce apoptosis in certain cells, including CD40, CD30, and FN14, also induce TNF and therefore work in a fashion similar to TNF-R2. Because TNF-R1 is the main signaling receptor for TNF, we now examine its activities in more detail. Binding of TNF to TNFR1 induces receptor oligomerization and recruitment of cytoplasmic signaling proteins, leading to the formation of the TNF-R1 signaling complex (TNF-RSC). The composition of the TNF-RSC and the following steps in TNF-R1–mediated signaling have been extensively studied over the last decade. Although further analysis will be required to discover all the players involved in this process, it is fair to say that to date, it is one of the best understood receptor signaling complexes and cascades in cell biology.
On activation of TNF-R1 by TNF-induced crosslinking at the plasma membrane, the TNFR1 DD serves as a docking site for the DD-containing adaptor protein TRADD. TRADD is recruited to the DD of TNF-R1 via a homotypic DD interaction. TRADD in turn recruits the TNF-R–associated factor-2 (TRAF2) and the receptorinteracting protein 1 (RIP1), a serine/threonine kinase. RIP1, however, can also directly bind to TNF-R1 via its own DD without the need for TRADD. The importance of this interaction remains unclear. Recruitment of TRAF2 (or TRAF5) by TRADD enables recruitment of cIAP1 and/or cIAP2 to the TNF-RSC. The ubiquitin ligase activities of both TRAFs and cIAPs are required for decoration of RIP1 by polyubiquitin chains and for NF-κB and MAP kinase activation. Ubiquitin chains can be formed via linkages of the ubiquitin subunits on different ε-amino groups of the seven different lysines present in ubiquitin or via the α-amino group at the amino-terminus of ubiquitin, with the latter creating linear ubiquitin chains. Thus far it is thought that polyubiquitin chains involved in TNF signaling are either linked via the ε-amino groups of lysine 63 (K63) or K48 of ubiquitin. However, recent data obtained by us and others revealed that linear ubiquitin chains also play an important role in this process. A protein complex termed LUBAC (for linear ubiquitin chain assembly complex) forms an integral part of the TNF-R1 signaling complex (Haas et al., 2009). Furthermore, LUBAC is required for

DEATH DOMAIN–CONTAINING RECEPTORS – DECISIONS BETWEEN SUICIDE AND FIRE |
31 |
TNF-R1 signaling complex
|
RIP1 |
TRADD |
|
|
TRAF2/5 |
TAB2 |
TAK1 |
|
TAB1 |
|
|
|
|
|
NEMO IKKβ |
|
cIAP1/2 |
|
|
|
IKKα |
|
|
NF-κB JNK p38
Gene induc on
Figure 3-3. Schematic representation of immunostimulatory, proinflammatory signaling by the TNF-R and DR3 systems. Binding of TNF and TL1A to their respective receptors leads to receptor trimerization and formation of a receptor signaling complex. First the adaptor protein TNF-R1–associated death domain (TRADD) is recruited via its DD to the DD of the receptor. TRADD then serves as an assembly platform for binding of TRAF2, cIAP1/2, and the receptor interacting kinase 1 (RIP1). TRAFs and cIAPs conjugate ubiquitin chains to various proteins in the complex, which allows for the recruitment of further signaling proteins, including the TAK/TAB and the IKK complexes, ultimately leading to activation of NF-κ B and the JNK and p38 MAP kinase pathways. See Color Plate 5.
efficient TNF-induced NF-κB activation because NF-κB essential modulator (NEMO) binds strongly to linear but only weakly to K63-linked ubiquitin chains. Together, K63-linked and linear polyubiquitination of different components of the TNF-RSC result in stable TNF-RSC formation and thereby enable the events that ultimately lead to activation of NF-κB and the JNK and p38 MAP kinase pathways. The molecular processes that lead to the activation of these pathways have been extensively reviewed in the past (Hayden and Ghosh, 2008; Wajant et al., 2003). The discovery of LUBAC as a novel integral component of the TNF-RSC and linear ubiquitination as a central player in the organization of this protein complex will undoubtedly substantially affect our current view of how these processes are regulated. It will be exciting to unravel these mechanisms at the molecular level and discover how they control the function of TNF.
3.1.2. TNF and TNF blockers in the clinic
Soon after the isolation of TNF, it became clear that the systemic administration of TNF is highly toxic as
a result of the extreme cytokine production it induces. This inflammation-like syndrome prevented the further development of TNF for systemic use. However, a technique called isolated limb perfusion (ILP) has been developed by Ferdinand Lejeune in Lausanne, Switzerland (Lejeune et al., 1995). ILP facilitates local and exclusive administration of TNF to, for example, an arm or leg of a patient with cancer. ILP with TNF in combination with chemotherapeutic drugs led to complete response rates in some patients with sarcomas and melanomas on extremities and showed improved penetrance of the cytostatic drugs melphalan and doxorubicin into tumors in animal models. The possibility to inhibit any leaked TNF in the rest of the body with therapeutic TNF blockers, which are now available, may help to overcome the limitations posed so far on ILP by the detrimental effects of potential TNF leakage. Interestingly, TNF specifically disrupts tumor-supporting blood vessels while sparing normal blood vessels. It would be interesting to examine whether endothelial cells in the tumor-associated neovasculature are particularly sensitive to TNF-induced apoptosis and what the biochemical
32 |
|
|
HENNING WALCZAK AND CHAHRAZADE KANTARI |
|
Primarily apopto c signaling systems |
Primarily immunos mulatory, proinflammatory |
|||
(CD95 and TRAIL systems) |
signaling systems (TNF and DR3 systems) |
|||
Complex I |
|
|
Complex I |
|
FADD |
RIP1 |
TRADD |
|
|
|
|
|||
Complex II |
|
|
TRAF2/5 |
Complex II |
Caspase- 8 |
TAB2 |
TAK1 |
|
|
|
TAB1 |
|
|
|
NEMO |
NEMOIKKβ |
|
cIAP1/2 |
|
|
IKKα |
|
|
|
NF-κB
MAPK
APOPTOSIS |
Gene induc on |
APOPTOSIS |
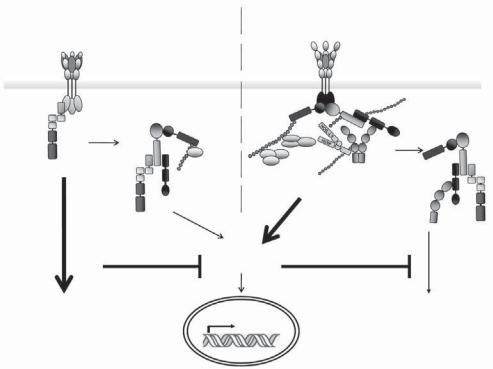
Figure 3-4. Complex I and complex II: spatial dissociation between proapoptotic and proinflammatory signaling in death receptor signal transduction. For both the CD95 and TRAIL systems, as well as the TNF and DR3 systems, the complex defined as complex I is the protein complex that forms at the plasma membrane and exerts the primary function of the respective receptor (i.e., apoptosis for CD95 and TRAIL-R1/R2 and gene induction via NF-κB and MAPK activation by TNFR-R1 and DR3). By an undetermined mechanism, the primary adaptor protein for the di erent complexes I dissociates from the DD of the respective receptor, together with a number of other proteins assembled in complex I (but without the receptor), and recruits additional proteins from the cytosol. This complex II then triggers the secondary function of each receptor. In the case of proapoptotic receptors, this is gene induction via activation of NF-κB and the MAP kinases pathways; in the case of the primarily immunostimulatory, proinflammatory receptors, it is induction of apoptosis. Successful completion of the respective primary signal interferes with the execution of the respective secondary signal. See Color Plate 6.
basis for this is. ILP is approved for unresectable soft tissue sarcoma, and it has also been successfully applied in the treatment of various other local tumors. The success of this technique proved that TNF can be used to treat cancer, albeit only when administration is locally restricted and by exerting its killing activity on an unexpected cellular target. Hence successful TNF treatment has mainly become a matter of targeted delivery to the tumor site.
A molecular way of achieving targeted delivery is to create fusion proteins in which TNF is conjugated to antibody fragments or natural ligands that specifically recognize surface proteins on tumor cells or in the tumor stroma. Using this technique, significant killing of tumor cells has been obtained, and exciting new recombinant proteins are currently being investigated. As an example, a melanoma-specific antibody conjugated to recombinant human TNF exhibited very good killing activity against TNF-resistant melanoma cells, both in
vitro and in vivo. Thus these types of fusion proteins or conjugates may not only result in more effective tumor targeting, but may also enhance the killing activity of TNF, most likely by providing membrane fixation and thereby enabling higher order receptor cross-linking on the target cell. Similar fusion proteins have also been constructed with CD95L and TRAIL. The preclinical results obtained with some of the proteins are very encouraging.
The by far most important clinical development in the TNF field to date emerged from the initially discouraging observation that TNF exerts an inflammatory response. When scientists started investigating the upside of this, they realized that interference with this response can be used to treat inflammatory conditions. In the beginning it was thought that sepsis could be targeted by inhibiting TNF. However, it was overlooked that Daniela Mannel¨ and her team, then in Heidelberg, Germany, had already shown that TNF plays an ambiguous