


APOPTOSIS AND HOMEOSTASIS IN THE EYE |
177 |
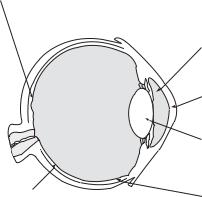
Optic nerve |
Iris |
|
Cornea
 Pupil
Pupil

 Macula
Macula
Lens
Retina




Iris
Figure 16-1. Anatomy of human eye. Reprinted courtesy of the National Eye Institute.
apoptosis are expressed in situ in the developing lens, classical apoptosis is not directly involved in denucleation of the lens epithelial cells or in lens development. Normally, apoptotic cells die and are phagocytosed by neighboring cells. By contrast, denucleation culminates in the differentiation of lens epithelial cells into long-lived lens fiber cells that remain throughout the individual’s life. During the conversion of the lens epithelial cells to lens fiber cells, the cytoplasm becomes homogenous and accumulates high concentrations of lens-specific proteins, called crystallins, which provide the lens fiber cells and the lens itself with a crystalclear transparency that is vital for normal vision. The crystallins constitute approximately 90% of the watersoluble proteins of the lens and provide it with its refractile properties.
The lens grows throughout life with the continuous programmed removal of nuclei and other organelles from lens fiber cells. It is noteworthy that in spite of this continuous cell growth and differentiation, spontaneous tumors of the lens (other than experimentally induced neoplasms) have not been described in any species except the cat.
1.2. Retina
Apoptosis plays a pivotal role in the development of the central nervous system (CNS) and the retina. Retinal ganglion cells (RGCs) project from the eye via the optic nerve to the visual centers in the brain. During retinal development, an overproduction of RGCs occurs, with approximately half of the RGCs dying after reaching the visual centers in the brain. One theory holds
that the peripheral neurons compete for a limited supply of neurotrophic factors and that apoptosis is employed to maintain a balance between growth factor supply and neuron demand. Evidence supporting this neurotrophin hypothesis stems from observations showing that removal of one eye promotes the survival of neurons projecting from the other eye to the visual centers in the brain. In rats, up to 90% of the RGCs die during the first postnatal week. In the chick eye, two distinct periods of apoptosis are invoked to shape the development of the RGC population. Apoptosis at an early stage of retinal development is believed to create space for incoming axons of RGC to form the optic nerve. At a later stage of retinal development, apoptosis of RGCs follows innervation and synapse formation with the visual centers in the brain. Emerging evidence suggests that transforming growth factor-β2 (TGF-β2) plays a central role in apoptosis of RGCs during retinal development.
2. ROLE OF APOPTOSIS IN DISEASES OF THE EYE
2.1. Glaucoma
It has been estimated that by the year 2020, almost 80 million people will have glaucoma, which is one of the leading causes of irreversible blindness. Glaucoma is a disease of the optic nerve. A common misconception is that glaucoma is produced by chronic elevation in intraocular pressure. Although glaucoma is often associated with increased intraocular pressure, a significant number of individuals experience glaucoma even though their intraocular pressures are within the normal ranges (i.e., normal pressure glaucoma). A significant body of evidence indicates that ocular hypertension alone is neither necessary nor sufficient for the production of optic neuropathy. It has been proposed that in some cases, glaucoma is an immune-mediated disease. Some glaucoma patients produce antibodies directed at heat shock proteins (HSPs), such as HSP60 and HSP27, which are upregulated in glaucomatous optic nerve heads. It has been proposed that HSPs have important neuroprotective properties, but under pathological conditions they can serve as immunogens that provoke adaptive immune responses that culminate in the generation of autoantibodies against retinal antigens. Antibodies directed against HSP27 have been detected in the sera of glaucoma patients, and when tested in vitro, these sera induce RGC death via an apoptotic mechanism. Animal studies have recently demonstrated that immunization with HSP27 and HSP60 results in optic neuropathy that is characterized by a T-cell infiltrate and apoptosis of

178 |
JERRY Y. NIEDERKORN |
RGC. T cells isolated from HSP60 and HSP27 immunized rats induced apoptosis of RGC cells in vitro by a process characteristic of apoptosis. Moreover, HSP-immune T cells elaborated a soluble factor that induced apoptosis of the Fas+ RGC cells in a cell contact–independent manner, which could be blocked with antibody specific for FasL. Additional studies demonstrated that recombinant human FasL alone induced apoptosis of rat RGC cells in vitro, suggesting that the generation of anti-RGC T cells was antigen-specific, but the effector mechanism that culminated in apoptosis of RGC cells was antigen nonspecific. Although these findings are provocative, they do not account for all cases of glaucoma, and competing hypotheses suggest that other conditions such as neurotrophin deprivation can be key factors in the pathogenesis of glaucoma.
2.2. Age-related macular degeneration
Age-related macular degeneration (AMD) is the leading cause of blindness in the elderly in developed countries and affects approximately 35% of individuals 75 years of age or older. AMD is characterized by progressive degeneration of the photoreceptors and retinal pigment epithelial (RPE) cells in the small central portion of the retina called the macula, which is responsible for high visual acuity (Figure 16-1). The growth of new blood vessels in the choroid layer (i.e., choroidal neovascularization; CNV), which lies beneath the retina, develops in 10% of AMD patients yet accounts for 90% of the blindness in AMD (Figure 16-2). Macrophages have been implicated in the etiology of AMD, with some studies suggesting that macrophages stimulate CNV, whereas other studies indicate that they inhibit CNV. Investigations in a mouse model of laser-indu- ced CNV have provided evidence that CNV is the result of an imbalance in FasL-mediated apoptosis of newly generated choroidal vascular endothelial cells. Evidence suggesting that FasL-induced apoptosis might regulate CNV has arisen from studies on ocular biopsies from AMD patients, which demonstrated that new choroidal blood vessels in AMD patients expressed Fas receptor. Investigations in a mouse model of laser-induced CNV indicated that CNV was significantly increased in mice with defective, nonfunctional FasL (gld/gld) and in Fas receptor-deficient mice (lpr/lpr). Studies from interleukin (IL)-10 knockout (KO) mice revealed that the absence of IL-10 resulted in polarization of intraocular macrophages to an antiangiogenic phenotype. As its name implies, AMD is a disease of senescence. Interestingly, the ability of laser-induced injury to stimulate CNV increases as mice age, and the level
Figure 16-2. Retinal neovascularization in age-related macular degeneration (AMD). A. Normal retina. B. Early AMD with retinal neovascularization. C. AMD with retinal neovascularization and degeneration of macula (arrow). Reprinted courtesy of the National Eye Institute. See Color Plate 16.
of IL-10 in the posterior compartment of the eye is highest in older mice. Moreover, FasL expression on macrophages diminishes with age. Macrophages from aged mice have reduced expression of FasL and demonstrate a diminished capacity to inhibit vascular endothelial cell growth. These findings have led to the hypothesis that resident macrophages in the eye are crucial for maintaining a normal level of choroidal vascularization, and that as we age, the level of FasL diminishes on ocular macrophages and the concentration of immunosuppressive cytokines, such as IL-10, in the intraocular milieu increases. Macrophage function is influenced by the microenvironment, and as such, macrophages can behave either as either proangiogenic or antiangiogenic effectors. However, in the aging eye, the combination of elevated IL-10 and diminished FasL tilt the ocular macrophage population to a proangiogenic phenotype.
APOPTOSIS AND HOMEOSTASIS IN THE EYE |
179 |
3. APOPTOSIS AND SURVEILLANCE OF
INTRAOCULAR TUMORS
The immune privilege of the eye has been recognized for more than 140 years and is known to permit the long-term survival of tissue and tumor allografts. However, some murine tumors transplanted into the anterior chamber circumvent immune privilege and undergo immune rejection in the eye. A number of theories have been offered to explain the circumvention of ocular immune privilege. Among these theories is the notion that some tumors succumb to apoptosis induced by cell membrane molecules that are expressed on intraocular cells. The cells lining the interior of the eye are decorated with a variety of molecules that disable immune effector elements and in some cases, can also induce apoptosis of intraocular tumor cells. These include FasL, tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), programmed death ligand-1 (PD-L1), and complement regulatory proteins. However, at least one of these molecules (TRAIL) can also purge the eye of tumor cells bearing its apoptosis-inducing receptor (DR5). TRAIL is expressed on a wide variety of ocular cells and is regulated by interferon-γ (IFN-γ), as its expression is virtually absent in the eyes of IFN-γ KO mice. Lee and coworkers reported that P815 tumor cells not expressing TRAIL receptor 2 (DR5) grew progressively in the eyes of allogeneic BALB/c mice. However, the same tumor cells underwent brisk rejection if they were transfected with TRAIL-R2 (DR5) cDNA to induce DR5 expression. Moreover, in vitro studies testing a variety of DR5+ tumor cell lines demonstrated that DR5+ tumor cells underwent apoptosis when incubated in vitro with ocular cells that constitutively expressed TRAIL. Apoptosis could be blocked with an antagonistic anti-TRAIL antibody, thereby confirming the specificity of the TRAILinduced tumor cell apoptosis by ocular cells. Although these results involved TRAIL-induced apoptosis of allogeneic and xenogeneic tumors, it is noteworthy that in a follow-up study, more than half of the human ocular melanoma cell lines tested expressed TRAIL-R2 and were susceptible to TRAIL-induced apoptosis. Thus human ocular tumors express TRAIL-R2 and are susceptible to TRAIL-induced apoptosis. It remains to be confirmed whether this constitutes an effective immune surveillance mechanism for intraocular tumors.
4. APOPTOSIS AND OCULAR IMMUNE PRIVILEGE
The immune privilege of the eye is the sum total of unique anatomical, physiologic, and immunoregulatory processes that block the induction and expression of
both innate and adaptive immune effector mechanisms. The blood vessels of the anterior segment of the eye are non-fenestrated and create a blood:ocular barrier that limits the extravasation of circulating leukocytes into the eye. Leukocytes that succeed in entering the anterior chamber encounter aqueous humor, which contains a potpourri of anti-inflammatory and immunosuppressive molecules. Among these is a 10-kDa peptide that induces apoptosis of natural killer cells, T cells, neutrophils, and macrophages. Leukocytes that escape apoptosis mediated by aqueous humor-borne factors are greeted by at least three different cell membrane-bound molecules – FasL, TRAIL, and PD-L1 – that are expressed on cells lining the interior of the eye and have been shown to induce apoptosis of activated T lymphocytes.
One of the remarkable manifestations of ocular immune privilege is the exceptionally high survival rate for corneal allografts. In uncomplicated first-time cases, corneal allografts experience a 90% acceptance rate, even though HLA-matching is not employed and systemic immunosuppressive drugs are not used. Studies in rodents have demonstrated that corneal cells express both FasL and PD-L1, which induce apoptosis of activated T lymphocytes and whose expression is crucial for corneal allograft survival.
The immune privilege of the anterior chamber (AC) of the eye is also maintained by a dynamic immunoregulatory process that downregulates T-cell–based inflammation in an antigen-specific manner. That is, antigens introduced into the AC of the eye induce an immune deviation that suppresses T-cell–mediated immunity such as DTH and cytotoxic T-lymphocyte (CTL) activity while preserving antibody responses. This anterior chamber-associated immune deviation (ACAID) is associated with the immune privilege of tumor allografts transplanted into the AC of the eye and corneal allografts transplanted over the AC. ACAID is a complex immunoregulatory phenomenon that is initiated when antigens are introduced into the AC and involves the eye, thymus, spleen, and sympathetic nervous system. FasL-induced apoptosis appears to be intimately involved in the induction of ACAID. Early evidence that FasL-induced apoptosis was involved in ocular immune privilege in general and ACAID in specific arose from experiments in which herpes simplex virus-1 (HSV-1) was injected into the AC of mice. Injection of HSV-1 into subcutaneous sites elicits robust HSV-specific DTH. However, HSV-1 injected into the AC induces ACAID, as demonstrated by the downregulation of HSV-1–specific DTH. However, AC injection of HSV-1 into mice with defective Fas receptor (lpr) or FasL (gld) not only fails to induce ACAID, but in fact, stimulates positive
180 |
|
|
|
|
|
|
|
|
|
|
|
|
JERRY Y. NIEDERKORN |
|
HSV-1–specific |
DTH |
and |
provokes |
Retinal macrophages express FasL |
|
|
||||||||
intense |
ocular |
inflammation. These |
and regulate retinal blood vessel |
|
|
|||||||||
development by inducing apoptosis of |
|
|||||||||||||
|
|
|
|
|
|
|
|
|
|
|
||||
results suggest that FasL-induced apop- |
of Fas+ vascular endothelial cells. |
|
|
|||||||||||
tosis is |
necessary for |
the |
induction of |
|
|
|
FasL-induced apoptosis of antigen |
|||||||
immune |
deviation |
(i.e., |
ACAID) |
and |
|
|
|
presenting cells and infiltrating |
||||||
|
|
|
lymphocytes promotes immune deviation |
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
||
that the absence of FasL/FasR interac- |
|
|
|
and immune privilege in anterior chamber. |
||||||||||
tions robs the anterior chamber of its |
|
|
|
|
||||||||||
immune privilege. This was confirmed |
|
|
|
FasL-induced apoptosis of |
||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
||
in |
additional |
experiments |
involving |
|
|
|
infiltrating lymphocytes preserves |
|||||||
hapten-derivatized spleen cells. |
Syn- |
|
|
|
corneal allograft survival. |
|||||||||
|
|
|
|
|||||||||||
geneic |
spleen |
cells |
treated |
with |
the |
|
|
|
Apoptosis-like process shapes lens |
|||||
hapten |
trinitrophenol |
(TNP-spl) |
will |
|
|
|
||||||||
|
|
|
development and allows continuous |
|||||||||||
induce TNP-specific DTH responses |
|
|
|
“growth” of lens throughout life. |
||||||||||
|
|
|
|
|||||||||||
when injected subcutaneously, whereas |
|
|
|
|
||||||||||
the |
same |
TNP-spl |
injected |
into |
the |
TRAIL is expressed throughout the |
Two waves of apoptosis shape |
|||||||
retinal ganglion cell development and |
||||||||||||||
AC |
induce inhibition |
of |
TNP-specific |
eye and induces apoptosis of |
|
|||||||||
|
morphogenesis of the retina. |
|||||||||||||
tumors expressing TRAIL-R2 (DR5). |
||||||||||||||
DTH. However, if spleen cells from Fas |
|
|||||||||||||
|
|
|
|
|||||||||||
receptor–deficient mice are derivatized |
Figure 16-3. Apoptosis shapes morphogenesis of the eye and sustains immune privilege |
|||||||||||||
by multiple mechanisms. Reproduced with permission from Nature Publishing Group; |
||||||||||||||
with TNP and |
injected |
into |
the AC, |
|||||||||||
Journal of Investigative Dermatology Symposium Proceedings 8:168, 2003. |
||||||||||||||
they fail to induce ACAID and instead |
||||||||||||||
|
|
|
|
|||||||||||
elicit TNP-specific DTH. Likewise, when |
|
maintenance of ocular immune privilege and in restrain- |
||||||||||||
TNP-derivatized |
spleen cells |
from |
normal mice are |
|||||||||||
ing ocular |
inflammation. However, failure of ocular |
|||||||||||||
injected into the eyes of FasL-deficient mice, ACAID |
||||||||||||||
immune privilege can have devastating consequences. |
||||||||||||||
is not induced, thereby confirming that FasL-induced |
||||||||||||||
Indeed, the three leading causes of infectious blind- |
||||||||||||||
apoptosis |
of TNP-derivatized |
cells |
is crucial for the |
|||||||||||
ness – trachoma, river blindness, and HSV keratitis – are |
||||||||||||||
induction of ACAID. Although these findings indicate a |
||||||||||||||
immune-mediated diseases in which the unrestrained, |
||||||||||||||
clear role for FasL-induced apoptosis in the induction of |
||||||||||||||
chronic immune response to ocular pathogens, rather |
||||||||||||||
ACAID, apoptosis induced by other means can also pro- |
||||||||||||||
than the direct cytopathic effects of the pathogens, is the |
||||||||||||||
mote the induction of ACAID. That is, TNP-derivatized |
||||||||||||||
primary cause of blindness. |
||||||||||||||
spleen cells from Fas receptor-deficient mice treated |
||||||||||||||
|
|
|
||||||||||||
with x-irradiation undergo apoptosis and when injected |
|
|
|
|||||||||||
into the AC, will induce ACAID as effectively as TNP- |
|
|
|
|||||||||||
derivatized spleen cells from wild-type mice. Follow-up |
SUGGESTED READINGS |
|
||||||||||||
studies demonstrated that apoptosis, elicited by either |
Apte RS, Richter J, Herndon J, Ferguson TA. Macrophages inhibit |
|||||||||||||
FasL or x-irradiation, stimulated the rapid production of |
||||||||||||||
neovascularization in a murine model of age-related macular |
||||||||||||||
IL-10, which altered the behavior of antigen-presenting |
||||||||||||||
degeneration. PLoS Medicine 2006;3(8):e310. |
||||||||||||||
cells and rendered them tolerogenic. |
|
|||||||||||||
|
Duenker N. Transforming growth factor-beta (TGF-beta) and |
|||||||||||||
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
programmed cell death in the vertebrate retina. International |
|||
5. CONCLUSIONS |
|
|
|
|
|
|
Review of Cytology 2005;245:17–43. |
|||||||
|
|
|
|
|
|
Ferguson TA, Apte RS. Angiogenesis in eye disease: immu- |
||||||||
|
|
|
|
|
|
|
|
|
|
|
||||
Apoptosis contributes to the development of the mam- |
nity gained or immunity lost? Seminars in Immunopathology |
|||||||||||||
malian eye in utero and is a crucial element in the |
2008;30(2):111–19. |
|
||||||||||||
homeostasis of the visual axis from birth to death (Fig- |
Ferguson TA, Griffith TS. A vision of cell death: insights |
|||||||||||||
ure 16-3). Although apoptosis is necessary for the main- |
into immune privilege. Immunological Reviews 1997;156:167– |
|||||||||||||
184. |
|
|
||||||||||||
tenance of vision, it also has a dark side and is a key |
|
|
||||||||||||
Galli-Resta L, Ensini M. An intrinsic time limit between gene- |
||||||||||||||
element in the pathogenesis of the two leading causes |
||||||||||||||
sis and death of individual neurons in the developing reti- |
||||||||||||||
of blindness, glaucoma and AMD. It is widely believed |
||||||||||||||
nal ganglion cell layer. Journal of Neuroscience 1996;16(7): |
||||||||||||||
that regulation of ocular inflammation is critical for pre- |
||||||||||||||
2318–24. |
|
|
||||||||||||
serving vision, as many ocular cells cannot regenerate, |
|
|
||||||||||||
Ishizaki Y, |
Jacobson MD, Raff MC. A role for caspases |
|||||||||||||
and immune-mediated injury to these cells would cul- |
||||||||||||||
in lens fiber differentiation. The Journal of Cell Biology |
||||||||||||||
minate in blindness. Apoptosis is a central process in the |
1998;140(1):153–8. |
|
||||||||||||
APOPTOSIS AND HOMEOSTASIS IN THE EYE |
181 |
Lee H-O, Herndon JM, Barreiro R, Griffith TS, Ferguson TA. TRAIL: A mechanism of tumor surveillance in an immune privileged site. Journal of Immunology 2002;169:4739–4744.
Linden R, Martins RA, Silveira MS. Control of programmed cell death by neurotransmitters and neuropeptides in the developing mammalian retina. Progress in Retinal and Eye Research
2005;24(4):457–91.
Niederkorn JY. The immune privilege of corneal grafts. Journal of Leukocyte Biology 2003;74(2):167–71.
Taylor AW. Ocular immunosuppressive microenvironment.
Chemical Immunology 2007;92:71–85.
Vrabec JP, Levin LA. The neurobiology of cell death in glaucoma. Eye (London, England) 2007;21 Suppl 1:S11– 14.
Wax MB, Tezel G, Yang J, Peng G, Patil RV, Agarwal N et al. Induced autoimmunity to heat shock proteins elicits glaucomatous loss of retinal ganglion cell neurons via activated T- cell-derived fas-ligand. Journal of Neuroscience 2008;28(46): 12085–96.
Yan Q, Liu JP, Li DW. Apoptosis in lens development and pathology. Differentiation; Research in Biological Diversity
2006;74(5):195–211.