

CONTRIBUTION OF APOPTOSIS TO PHYSIOLOGIC REMODELING OF THE ENDOCRINE PANCREAS |
203 |
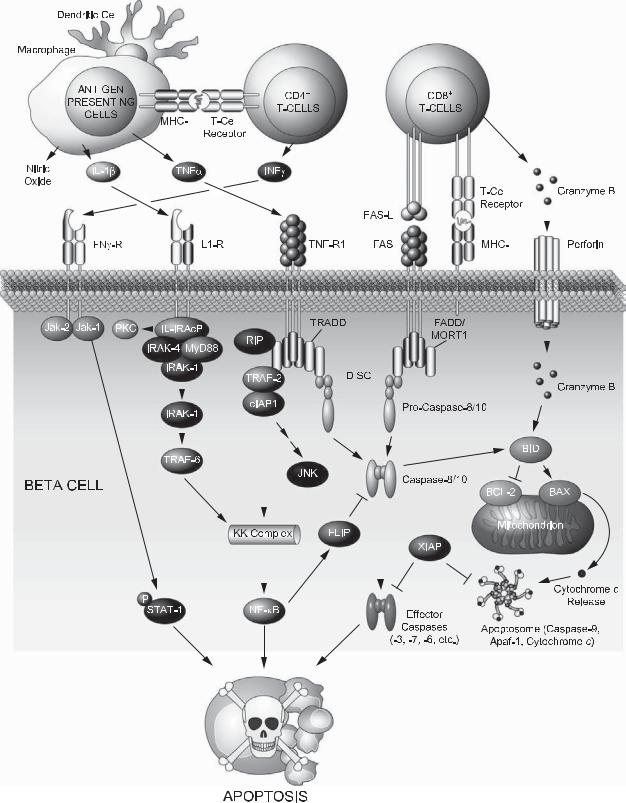
be limited to, peptides from insulin/proinsulin, glutamic acid decarboxylase, and the tyrosine phosphataselike protein IA-2.61 Na¨ıve beta cell–specific autoreactive CD4+ T cells that have escaped thymic negative selection become activated on encountering their cognate antigens presented by major histocompatibility complex (MHC) molecules on the surface of APCs in the pancreatic lymph nodes. On Initial activation, CD4+ T cells migrate through the pancreas, where they reencounter their antigen, become activated, and remain in the islets (insulitis). Activated CD4+ T cells produce interleukin (IL)-2 and interferon (IFN)-γ, further stimulating APCs to secrete nitric oxide (NO), IL-1β, and tumor necrosis factor (TNF)-α (Figure 19-1). TNF-α and IFN-γ stimulate secretion of chemokines from resident macrophages and endothelial cells, which help recruit CD8+ T cells.62 The latter secrete granules that contain perforin and the proapoptotic serine protease granzyme B. Perforin is a pore-forming protein63 that on release generates membrane-penetrating tubular structures that allow the delivery of granzyme B to beta cells64 (Figure 19-1). Granzyme B substrates include BID and caspase-3 and -7. Furthermore, CD8+ T cells trigger the extrinsic pathway of apoptosis by engaging FAS on the surface of beta cells.
Evasion of peripheral tolerance in T1D is influenced by genetic predisposition and environmental factors. For example, genome association studies have identified several candidate loci for T1D, among which are the HLA locus encoding MHC class I and II molecules required for antigen presentation and components of immune signaling pathways.65,66,67 This suggests that genetic predisposition may cause an exaggerated immune response, increasing the probability of autoreactive T-cell activation. In addition to genetic predisposition, environmental factors such as viral infection and toxins may also contribute to the activation of beta cell–reactive T cells.68 For example, virally infected beta cells may produce cytokine/chemokines that help recruit and activate lymphocytes.69 Furthermore, exposure of viral antigens on the surface of infected beta cells may activate autoreactive T cells by molecular mimicry of beta cell antigens.70
3.2. Mechanisms of beta cell death in type 1 diabetes
Within the inflammatory cytokine milieu during the progression of T1D, the destruction of beta cells is believed to be apoptotic in nature.71,72,73 Beyond the autoimmune-mediated damage, beta cells also participate in their own demise by secreting inflammatory cytokines, chemokines, and NO.74,75 Loss-of-function
studies in mouse models of the disease have further revealed that interfering with the FAS/FASL, perforin/granzyme B, and signal transduction downstream of the inflammatory cytokines confers protection from T1D.76,77,78,79,80,81,82 However, protection is chiefly partial, indicating that these mechanisms work in concert. An overview of apoptotic pathways implicated in T1D is provided in the following sections.
3.2.1. Apoptosis signaling pathways downstream of death receptors and inflammatory cytokines
Direct contact of beta cells and T lymphocytes leads to activation of the extrinsic apoptotic pathway downstream of FAS and TNF receptor (TNFR), ultimately culminating in activation of caspase-8 and -10.83 The subsequent transduction of the apoptotic signal is governed by protein–protein interaction domains that help assemble death-inducing signaling complexes (DISCs) on the cytoplasmic site of the death receptors84 (Figure 19-1). Within the DISCs, receptors, adaptors, and pro-caspase- 8 or -10 are assembled through selective homotypic engagement of protein–protein domains. These include the death domain (DD), the death effector domain (DED), and the caspase activation and recruitment domain (CARD) found in pro-caspases.85 Structural studies have revealed that the three-dimensional structure of these protein interaction domains are remarkably similar.86 The adaptor components of the DISCs vary depending on the death receptor. For example, in the case of FAS, the DISC is composed of FAS-associated DD-containing adaptor (FADD/MORT1) that engages the receptor through its DD and recruits pro-caspase- 8 or -10 through its DED domain (Figure 19-1). On the other hand, TNFR-associated DD-containing adaptor TRADD lacks DEDs and recruits pro-caspases indirectly through FADD (Figure 19-1). The DISCs serve as molecular platforms for caspase activation and subsequent activation of effector caspases-3, -6, and -7.87,88 In addition to caspase-driven proteolytic destruction, a mitochondrial amplification loop is also operative in beta cells whereby the proapoptotic molecule BID is cleaved by caspase-8 to initiate a program of mitochondrial dysfunction, release of cytochrome c, and further activation of the caspase cascade.89,90 (Figure 19-1). Consistent with these observations, BID-deficient beta cells are protected from FASand TNF-α–induced apoptosis.89 Furthermore, over-expression of BCL-2 protects human beta cells from apoptosis induced by inflammatory cytokines.91
Interference with FAS and TNFR signaling in certain settings protects against T1D. For example, mice
204 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
NIKA N. DANIAL |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Figure 19-1. Signaling pathways leading to beta cell destruction in type 1 diabetes. Image courtesy of Eric Smith of Dana-Farber Cancer Institute. See text for details. See Color Plate 21.
CONTRIBUTION OF APOPTOSIS TO PHYSIOLOGIC REMODELING OF THE ENDOCRINE PANCREAS |
205 |
over-expressing the dominant-negative form of FADD that binds the receptor but lacks DED76 or transgenic animals expressing the soluble TNF decoy receptor lacking a transmembrane domain92 are more resistant to T1D. The efficiency of such genetic maneuvers may further lie on the simultaneous inhibition of FAS and TNFR, given that they share signaling intermediates such as FADD. This is especially relevant as beta cell–specific deletion of FAS alone was not sufficient to block disease progression.93 Other strategies to preserve beta cell mass by blocking death receptor signaling include FLIP (FLICE/caspase-8 inhibitory protein)94,95,96 (Figure 19-1) and over-expression of the serine protease inhibitor CrmA, which inhibits caspase-8 and -10.97
Accumulating evidence has recently suggested that components of death receptor signaling may have a nonapoptotic role beneficial to beta cell mass. These functions may in turn be developmentally regulated and/ or dictated by distinct signaling complexes. For example, caspase-8 is required for physiologic beta cell growth45 and glucose-stimulated insulin secretion.98 Furthermore, differential recruitment of FLIP to caspase-8 at the cytoplasmic tail of FAS or selective association of DISC components in complexes devoid of FAS may carry nonapoptotic roles, including proliferation.98,99 Likewise, signaling downstream of TNFR1 can trigger apoptosis or proliferation, depending on the composition of complexes containing DD and DED proteins.100 In addition to DISC, TNF-α can induce the assembly of a complex that contains TRADD, TNFR-associated protein-2 (TRAF-2), receptor-associated kinase RIP, and cellular inhibitor of apoptosis proteins cIAP1 and cIAP2,101,102 which enables activation of nuclear factor kappa B (NF- κB).103,104 Depending on the cellular context, signaling downstream of NF-κB may impart proor antiapoptotic signals. For example, FLIP is an NF-κB target gene that inactivates the apoptosis arm of TNF-α signaling by inhibiting caspase-8.105,106 However, under other settings, NF-κB activation is chiefly a proapoptotic signal in beta cells.107 In light of these observations, any strategy devised to target death receptor signaling as a therapeutic approach in beta cell mass preservation must be selective for the apoptotic arm of the signaling cascade without interfering with proliferative signals emanating from the same receptor.
The effect of IL-1β on beta cell survival/death depends on dose and duration of exposure. Acute exposure to IL-1β stimulates insulin secretion108 and beta cell proliferation,109 allowing beta cells to compensate for insulin demand during inflammatory stress. However, in T1D, hyperglycemia prompts beta cells to secrete more IL-1β.110 Chronic activation of signals emanating
from the IL-1 receptor (IL-1R) interferes with mitochondrial metabolism,111 attenuates insulin secretion,112 and induces apoptosis.113 In this context, blocking IL-1β protects beta cells from apoptosis114,115 and ablation of IL1R preserves beta cell mass in experimental models of T1D,81 either through immunomodulation and/or direct effects on beta cells. Beta cells express a low-affinity IL1R1 that transduces the IL-1β signal and a high-affinity IL-1R2 that serves as a decoy receptor.116 On ligand binding, a multiprotein complex assembles at the IL1R that is composed of an accessory protein (IL-1RAcP) and two serine/threonine kinase IRAK-1 and -4, which are recruited to the receptor by the myeloid differentiation factor (MyD88)117 (Figure 19-1). Within this signaling complex, IRAK-4 phosphorylates IRAK-1, which then dissociates from the complex and binds TRAF6,118 leading to activation of IKK-NF-κB signaling axes.119 Upregulation of several NF-κB target genes such as iNos and Fas and downregulation of Bcl-2 compromises beta cell survival downstream of IL-1β signaling.75,120 Consistent with these observations, blocking NF-κB activation by transgenic expression of a nondegradable form of iκBα (the NF-κB super repressor) preserves beta cell mass and protects against T1D.121
IFN-γ often acts synergistically with IL-1β and TNF- α in apoptotic demise of beta cells.122 On engagement of its cognate receptor, IFN-γ activates the receptorassociated Janus kinase (JAK)-1 and -2, which subsequently phosphorylate select tyrosine residues on the cytoplasmic tail of the IFN-γ receptor (Figure 19-1). The signal transducer and activator of transcription (STAT)- 1 docks on the phosphorylated receptor, becomes itself target of tyrosine phosphorylation, and dimerizes to translocate to the nucleus123 (Figure 19-1). This signaling pathway is under negative regulation by inhibitors of cytokine signaling such as suppressors of cytokine signaling (SOCS)-1 and -3, which dock to the receptor, blocking activation of JAKs and access of STAT-1 to the receptor.124,125 The effect of IFN-γ on beta cell death is mediated through STAT-1126; however, the precise nature of proapoptotic genes regulated by STAT-1 in these cells is not known.120 Loss of STAT-1 function or gain of SOCS- 1/-3127,128,129,130,131 function in beta cells preserves their viability in the presence of inflammatory cytokines and protects against T1D. The benefits of inhibiting JAKSTAT signaling in this case are two-fold: direct preservation of beta cell survival and immunomodulation.
3.2.2. Oxidative stress
Chronic intra-islet inflammation during progression of T1D can also activate the intrinsic pathway of