

20 |
JASON B. GARRISON, ANDREAS KRIEG, KATE WELSH, YUNFEI WEN, AND JOHN C. REED |
include participation of regulatory microRNAs in some cases. Jafrac-2, in contrast, is a thioredoxin peroxidase found in the lumen of the endoplasmic reticulum. The N-terminus of Jafrac-2 is proteolytically removed upon import into the endoplasmic reticulum, thus exposing the IBM. 3D structures of Drosophila IBMs bound to BIRs of DIAPs have demonstrated an evolutionarily conserved mechanism of molecular recognition that is highly similar to that of their mammalian counterparts.
Chemical compounds have been generated with intent to mimic the tetrapeptide sequences of SMAC and related IAP antagonists for use as apoptosis sensitizers for cancer treatment. Both monovalent and bivalent compounds have been described, the latter binding two adjacent BIRs on target proteins such as XIAP (e.g, BIR2 and BIR3). Compounds with nanomolar potency have advanced to human clinical trials.
11. IAPS AND DISEASE
Somatic mutations and hereditary polymorphisms in IAP family genes have been associated with diseases. For example, chromosomal translocations involving c-IAP2 are found commonly in patients with non-Hodgkin’s lymphomas that arise at mucosal locations (mucosaassociated lymphoid tissue [MALT] lymphomas, or MALTomas). These translocations fuse portions of the c-IAP2 gene on chromosome 11 with portions of the MALT/paracaspase gene on chromosome 18, resulting in dysregulation of the signal transduction and E3 ligase activities of c-IAP2. Dysregulation of NF-κB signaling by c-IAP2/MALT oncoproteins requires TRAF2 binding to the BIR1 domain of c-IAP2. Moreover, amplification of the gene locus that contains c-IAP1 and c-IAP2 (which are located adjacent to each other in the human genome) has also been described in lung, cervical, and esophageal cancers.
Humans with X-linked lymphoproliferative disorder (XLP) have been identified with XIAP gene mutations that lead to defective expression of this IAP family member. Lymphocytes from patients with XLP have no detectable XIAP expression and demonstrate enhanced apoptosis in response to various apoptotic stimuli. The lymphoproliferative (rather than immunodeficient) phenotype seen in XIAP-deficient XLP patients appears, in part, to result from the observed low numbers of natural killer T-lymphocyte (NKT) cells, suggesting that XIAP may have a role in promoting NKT cell development or survival in humans.
Polymorphisms in the NAIP gene on human chromosome region 5q13.1 were initially linked to spinal muscular atrophy, a degenerative disease of spinal motoneu-
rons. However, more recent data suggest that the adjacent and partially overlapping SMN gene may be the culprit. In mice, polymorphisms in the Naip locus are associated with differential sensitivity to certain types of intracellular bacteria, particularly Legionella pneumophila, the cause of Legionnaires’ disease in humans. Most inbred strains of mice are resistant to infection, correlating with the non-permissiveness of macrophages to Legionella replication. It remains to be determined whether polymorphisms of the Naip gene in humans similarly impact susceptibility to bacterial infections.
In addition to polymorphic variants and gene mutants, altered expression of certain IAPs has been associated with diseases in the absence of any known change at the DNA level. In this regard, several IAPs become over-expressed in human cancers, sometimes correlating with clinical outcome. Expression of Survivin is frequently deregulated in cancer, representing one of the most common expression differences observed between malignant and normal tissues. Normally, Survivin expression is limited to fetal and embryonic tissues and is undetectable in fully differentiated adult tissues. Pathological over-expression of Survivin has been reported in most cancers, including breast, colorectal, esophageal, gastric, bladder, ovarian, stomach, pancreatic, liver, and uterine carcinoma, as well as lymphoma and neuroblastoma. High levels of Survivin protein in colorectal and esophageal cancer and glioma are associated with poor clinical outcome, treatment failure, or risk for relapse after therapy. Similarly, overexpression of various members of the IAP family has been documented in a variety of cancers and leukemias, including acute myelogenous leukemias (AML), renal cell carcinoma, prostate cancer, ovarian cancer, colon cancer, melanoma, and non–small-cell lung cancer. In some cases, expression of IAP family members such as XIAP, c-IAP1, or c-IAP2 has been correlated with differences in patient response to chemotherapy or patient survival. Significantly, more than one IAP may be overexpressed simultaneously in cancers, an observation with important implications for developing therapeutic strategies based on IAP antagonism.
Experimentally reducing (knockdown) IAP expression in various cultured cancer cells and tumor cell lines has provided evidence of their importance in sustaining survival of several types of malignant cells in some contexts. In vitro experiments with peptidyl inhibitors of IAPs based on endogenous antagonists such as SMAC and synthetic compounds that attack IAPs have also helped to validate certain members of the family as cancer drug discovery targets. Preclinical animal studies have suggested favorable therapeutic index and have
INHIBITOR OF APOPTOSIS PROTEINS |
21 |
demonstrated in vivo antitumor activity for such agents. To date, several SMAC-mimicking small-molecule compounds and two AS-ODN drugs are in human clinical trials for cancer.
In addition to cancer, in which IAP expression is commonly elevated, changes in expression of specific IAPs have been linked to neurodegenerative diseases. In Alzheimer’s disease (AD), expression of NAIP is decreased in cells taken from the human entorhinal cortex and inversely correlated with paired helical filament-11 (PHA-1) levels, a marker of neurofibrillary tangle pathology. The expression of XIAP is reportedly increased rather than decreased in human entorhinal cortex and hippocampal regions of AD patients, suggesting an attempted compensatory mechanism for surviving AD pathology. Experimental elevations in the expression of NAIP, XIAP, c-IAP1, or c-IAP2 protect neonatal motor neurons from axotomy. The ratio between XIAP and XAF-1 has been correlated with adult motor neuron protection and neonatal neuron sensitivity to axonal injury. NAIP and XIAP both appear to be necessary for preservation of motor neuron survival in rats after sciatic nerve axotomy. Interestingly, expression of XIAP increases in neurons after sciatic nerve injury, suggesting that it contributes to an endogenous defense mechanism. Studies in which neurons were microinjected with cytochrome c have demonstrated an important role for XIAP in preservation of cell survival. These and other related studies have implied that in postmitotic neurons, survival is possible even after mitochondria discharge their contents, provided that IAPs are available to suppress caspases.
Overall, our knowledge of the roles of IAPs in disease remains very limited. Subsequent chapters provide additional details about what is known thus far.
SUGGESTED READINGS
Chai J, Shiozaki E, Srinivasula SM, Wu Q, Dataa P, Alnemri ES, Shi Y. (2001) Structural basis of caspase-7 inhibition by XIAP.
Cell 104:769–80.
Conte D, Liston P, Wong JW, Wright KE, Korneluk RG. (2001) Thymocyte-targeted overexpression of XIAP transgene disrupts T lymphoid apoptosis and maturation. Proc Natl Acad Sci U S A 98:5049–54.
Conze DB, Albert L, Ferrick DA, Goeddel DV, Yeh WC, Mak T, Ashwell JD. (2005) Posttranscriptional downregulation of c- IAP2 by the ubiquitin protein ligase c-IAP1 in vivo. Mol Cell Biol 25:3348–56.
Dan HC, Sun M, Kaneko S, Feldman RI, Nicosia SV, Wang HG, Tsang BK, Cheng JQ. (2004) Akt phosphorylation and stabilization of X-linked inhibitor of apoptosis protein (XIAP).
J Biol Chem 279:5405–12.
Deveraux QL, Reed JC. (1999) IAP family proteins – suppressors of apoptosis. Genes Dev 13:239–52.
Deveraux QL, Roy N, Stennicke HR, Van Arsdale T, Zhou Q, Srinivasula SM, Alnemri ES, Salvesen GS, Reed JC. (1998) IAPs block apoptotic events induced by caspase-8 and cytochrome c by direct inhibition of distinct caspases. EMBO J 17:2215– 23.
Eckelman BP, Salvesen GS, Scott FL. (2006). Human inhibitor of apoptosis proteins: why XIAP is the black sheep of the family.
EMBO Rep 7:988–94.
Gyrd-Hansen M, Darding M, Miasari M, Santoro MM, Zender L, Xue W, Tenev T, da Fonseca PC, Zvelebil M, Bujnicki JM, Lowe S, Silke J, Meier P. (2008) IAPs contain an evolutionarily conserved ubiquitin-binding domain that regulates NF-kappaB as well as cell survival and oncogenesis. Nat Cell Biol 10: 1309–17.
Harlin H, Reffey SB, Duckett CS, Lindsten T, Thompson CB. (2001) Characterization of XIAP-deficient mice. Mol Cell Biol
21:3604–8.
Hinds MG, Norton RS, Vaux DL, Day CL. (1999) Solution structure of a baculoviral inhibitor of apoptosis (IAP) repeat. Nat Struct Biol 6:648–51.
Hunter AM, LaCasse EC, Korneluk RG. (2007) The inhibitors of apoptosis (IAPs) as cancer targets. Apoptosis 12:1543– 68.
Joazeiro CA, Weissman AM. (2000) RING finger proteins: mediators of ubiquitin ligase activity. Cell 102:549–52.
Lu M, Lin SC, Huang Y, Kang YJ, Rich R, Lo YC, Myszka D, Han J, Wu H. (2007) XIAP induces NF-kappaB activation via the BIR1/TAB1 interaction and BIR1 dimerization. Mol Cell
26:689–702.
Morgan J, Yin Y, Borowsky A (1999) Breakpoints of the t(11;18)(q21;q21) in mucosa-associated lymphoid tissue (MALT) lymphoma lie within or near the previously undescribed gene MALT1 in chromosome 18. Cancer Res 59: 6205–13.
O’Connor DS, Grossman D, Plescia J, Li F, Zhang H, Villa A, Tognin S, Marchisio PC, Altieri DC. (2000) Regulation of apoptosis at cell division by p34cdc2 phosphorylation of survivin.
Proc Natl Acad Sci U S A 97:13103–17.
Oost TK, Sun C, Armstrong RC, Al-Assaad AS, Betz SF, Deckwerth TL, Ding H, Elmore SW, Meadows RP, Olejniczak ET, Oleksijew A, Oltersdorf T, Rosenberg SH, Shoemaker AR, Tomaselli KJ, Zou H, Fesik SW. (2004) Discovery of potent antagonists of the antiapoptotic protein XIAP for the treatment of cancer. J Med Chem 26:4417–26.
Perrelet D, Ferri A, Liston P, Muzzin P, Korneluk RG, Kato AC. (2002) IAPs are essential for GDNF-mediated neuroprotective effects in injured motor neurons in vivo. Nat Cell Biol 4:175–9.
Reed JC. (2001) The Survivin saga goes in vivo. J Clin Invest
108:965–9.
Santoro MM, Samuel T, Mitchell T, Reed JC, Stainier DY. (2007) Birc2 (cIap1) regulates endothelial cell integrity and blood vessel homeostasis. Nat Genet 39:1397–1402.
Srinivasula SM, Ashwell JD (2008) IAPs: What’s in a name? Mol Cell 30:123–35.
22 |
JASON B. GARRISON, ANDREAS KRIEG, KATE WELSH, YUNFEI WEN, AND JOHN C. REED |
Temesgen S, Welsh K, Lober T, Togo SH, Zapata JM, Reed JC. (2006) Distinct BIR domains of cIAP1 mediate binding to and ubiquitinylation of tumor necrosis factor receptor-associated factor 2 and second mitochondrial activator of caspases. J Biol Chem 281:1080–90.
Wang Z, Cuddy M, Samuel T, Welsh K, Schimmer A, Hanaii F, Houghten R, Pinilla C, Reed JC. (2004) Cellular, biochemi-
cal, and genetic analysis of mechanism of small molecule IAP inhibitors. J Biol Chem 279:48168–76.
Ziegler DS, Wright RD, Kesari S, Lemieux ME, Tran MA, Jain M, Zawel L, Kung AL. (2008) Resistance of human glioblastoma multiforme cells to growth factor inhibitors is overcome by blockade of inhibitor of apoptosis proteins. J Clin Invest
118:3109–22.
3Death Domain–Containing Receptors – Decisions between Suicide and Fire
Henning Walczak and Chahrazade Kantari
1. INTRODUCTION
The tumor necrosis factor (TNF) was among the first cytokines to be characterized both functionally and molecularly. This is due to its pivotal role in physiology and also in pathological conditions. A major driving force behind the initial studies into TNF and its functions was the hope its name already spells out: could TNF serve as a new drug to treat cancer? However, in the end TNF inhibition turned out to be an extremely successful novel therapeutic concept that has revolutionized the treatment of inflammatory diseases such as rheumatoid arthritis, Crohn’s disease, and psoriasis, a truly remarkable turnaround.
This turnaround, however, brings up the question of whether there is a molecular basis for what appears to be a misconception at the onset of TNF research. Or is it possible that there is no misconception, but that the two apparently contradictory outcomes are merely the result of different functions of a versatile cytokine? The answer lies in the molecular understanding of the signaling pathways triggered by TNF itself, as well as by the other members of a family of proteins we today refer to as the TNF superfamily (TNFSF) of cytokines. The two fundamental cellular responses triggered by this family of cytokines (i.e., the induction of cell death by apoptosis and the triggering of inflammation) are encrypted in these pathways. They are triggered when TNFSF cytokines cross-link their cognate receptors. By examining the molecular signaling pathways engaged by receptors capable of inducing cell death, we shed light on the differences and similarities between the signals that lead to these diametrically different outcomes.
Receptors capable of inducing cell death by apoptosis form part of the TNF receptor superfamily (TNFRSF). Membership in this protein family is endowed by presence of up to six repeats of a characteristic cysteinerich domain (CRD) in the extracellular portion of the receptor. These CRDs are crucial for each receptor’s binding of its cognate ligand. TNFRSF members are receptors that are able to induce a variety of biological functions after cross-linking by their respective ligands. The versatility of their response spans from the induction of cell death by apoptosis to proliferation and from down-modulation of an immune response to immunostimulatory, proinflammatory action. Most members of the TNFRSF are type I transmembrane proteins, but some are only attached to the plasma membrane by a glycosylphosphatidylinositol (GPI) anchor. Other TNFRSF members are secreted soluble molecules, acting as decoys for their respective ligands. To date, the TNFRSF consists of 30 members, and the death receptors form a subgroup of this family.
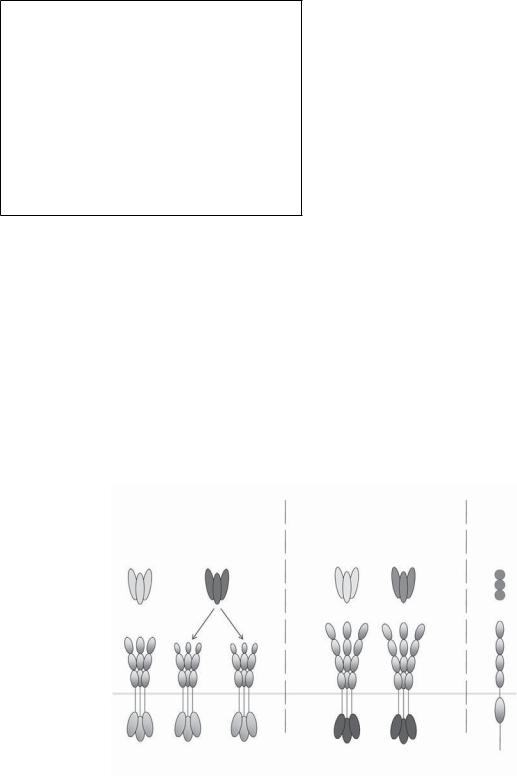
Death receptors are characterized by the presence of an intracellular death domain (DD), a stretch of approximately 80 amino acids that adopts a characteristic conformation, now generally referred to as the DD fold. To date, six human DD-containing receptors have been identified: TNF-R1 (p55/p60 TNF-R), CD95 (Fas, APO- 1), death receptor 3 (DR3, TRAMP), TRAIL-R1 (DR4), TRAIL-R2 (DR5), and DR6 (TNFRSF21). These receptors are activated by their respective ligands: TNF, CD95L (FasL/APO-1L), TL1A, TRAIL (Apo2L), and, as recently suggested, a specific amino-terminal cleavage fragment of the β-amyloid precursor protein (APP), N-APP (Table 3-1 and Figure 3-1). As expected, the DD plays a crucial role in signaling induced by these receptors as
23
24 |
HENNING WALCZAK AND CHAHRAZADE KANTARI |
Table 3-1. The six human DD-containing receptors and their ligands
Receptor(s) |
Ligand(s) |
|
|
TNF-R1 (p55/p60 TNF-R) |
TNF |
|
lymphotoxin-α (LT-α) |
CD95 (Fas/APO-1) |
CD95L (FasL/APO-1L) |
DR3 (TRAMP) |
TL1A |
TRAIL-R1 (DR4) |
TRAIL (Apo2L) |
TRAIL-R2 (DR5) |
|
DR6 |
APP (not a member of the TNFSF; |
|
possibly another ligand is still |
|
not identified) |
it enables the recruitment of proteins that themselves contain a DD. The most prominent and decisive integrators of death receptor signaling are the proteins known as Fas-associated DD (FADD or MORT1) and TNFRassociated DD (TRADD). FADD and TRADD are adaptor proteins. This means they themselves do not exert any enzymatic function but instead act by forming a bridge between proteins, in this case between receptor and signaling effector proteins. Dependent on whether a death receptor recruits FADD or TRADD to its DD, it can be categorized into one of two classes: although cross-
Primary signal:
Apoptosis
CD95L TRAIL
linking of CD95, TRAIL-R1, and TRAIL-R2 results in the recruitment of FADD, TRADD is recruited to ligandactivated TNF-R1 and DR3. The FADD-recruiting class of death receptors induces apoptosis as their primary signaling output, whereas the TRADD binders primarily induce immunostimulatory, proinflammatory signaling. At this time the only DD-containing receptor that does not fall into any of these two categories is DR6. It will be interesting to discover which third kind of signaling may be triggered by this receptor. A first hint comes from the recent identification of N-APP as a DR6 ligand.
In the following sections, we cover the five different death receptor–ligand systems. We first address the two FADD-recruiting receptor-ligand systems, the CD95 and the TRAIL systems. In this part we also make reference to some of the historical landmark findings in apoptosis research originating from the study of these systems. In the following section, we discuss the TRADDrecruiting immunostimulatory, proinflammatory TNF-R1 and DR3. Then we examine the current knowledge on DR6 before we finally juxtapose the signaling systems of FADDand TRADD-binding receptors to define the similarities and differences between the two main types of signaling triggered by DD-containing receptors.
Primary signal: Immune s mula on (Inflamma on)
TNF |
TL1A |
N- APP |
|
|
? |
CD95 |
TRAIL-R1 |
TRAIL-R2 |
TNFR1 |
DR3 |
DR6 |
(APO -1/Fas) |
(D R 4) |
(D R 5) |
|
(TRAMP) |
|
Figure 3-1. The human death domain-containing receptors and their known ligands. The six human death domain (DD)–containing receptors are transmembrane proteins that contain repeats of two to four cysteinerich domains (CRDs) in the extracellular portion required for the ligation of their cognate ligand and an intracellular DD capable of recruiting adaptor proteins that will trigger downstream intracellular signals (light gray for CD95/TRAIL systems and dark gray for TNF-R/DR3 systems). DD-containing receptors mainly trigger two signals: CD95, TRAIL-R1, and TRAIL-R2 interact with the adaptor protein FADD and therefore induce apoptosis as their primary signaling output, whereas TNFR-1 and DR3 interact with TRADD and hence mainly trigger proinflammatory signals. The molecular events that initiate DR6 signaling have not been elucidated.