

- •Contents
- •Contributors
- •Part I General Principles of Cell Death
- •1 Human Caspases – Apoptosis and Inflammation Signaling Proteases
- •1.1. Apoptosis and limited proteolysis
- •1.2. Caspase evolution
- •2. ACTIVATION MECHANISMS
- •2.2. The activation platforms
- •2.4. Proteolytic maturation
- •3. CASPASE SUBSTRATES
- •4. REGULATION BY NATURAL INHIBITORS
- •REFERENCES
- •2 Inhibitor of Apoptosis Proteins
- •2. CELLULAR FUNCTIONS AND PHENOTYPES OF IAP
- •3. IN VIVO FUNCTIONS OF IAP FAMILY PROTEINS
- •4. SUBCELLULAR LOCATIONS OF IAP
- •8. IAP–IAP INTERACTIONS
- •10. ENDOGENOUS ANTAGONISTS OF IAP
- •11. IAPs AND DISEASE
- •SUGGESTED READINGS
- •1. INTRODUCTION
- •2.1. The CD95 (Fas/APO-1) system
- •2.1.1. CD95 and CD95L: discovery of the first direct apoptosis-inducing receptor-ligand system
- •2.1.2. Biochemistry of CD95 apoptosis signaling
- •2.2. The TRAIL (Apo2L) system
- •3.1. The TNF system
- •3.1.1. Biochemistry of TNF signal transduction
- •3.1.2. TNF and TNF blockers in the clinic
- •3.2. The DR3 system
- •4. THE DR6 SYSTEM
- •6. CONCLUDING REMARKS AND OUTLOOK
- •SUGGESTED READINGS
- •4 Mitochondria and Cell Death
- •1. INTRODUCTION
- •2. MITOCHONDRIAL PHYSIOLOGY
- •3. THE MITOCHONDRIAL PATHWAY OF APOPTOSIS
- •9. CONCLUSIONS
- •SUGGESTED READINGS
- •1. INTRODUCTION
- •3. INHIBITING APOPTOSIS
- •4. INHIBITING THE INHIBITORS
- •6. THE BCL-2 FAMILY AND CANCER
- •SUGGESTED READINGS
- •6 Endoplasmic Reticulum Stress Response in Cell Death and Cell Survival
- •1. INTRODUCTION
- •2. THE ESR IN YEAST
- •3. THE ESR IN MAMMALS
- •4. THE ESR AND CELL DEATH
- •5. THE ESR IN DEVELOPMENT AND TISSUE HOMEOSTASIS
- •6. THE ESR IN HUMAN DISEASE
- •7. CONCLUSION
- •7 Autophagy – The Liaison between the Lysosomal System and Cell Death
- •1. INTRODUCTION
- •2. AUTOPHAGY
- •2.2. Physiologic functions of autophagy
- •2.3. Autophagy and human pathology
- •3. AUTOPHAGY AND CELL DEATH
- •3.1. Autophagy as anti–cell death mechanism
- •3.2. Autophagy as a cell death mechanism
- •3.3. Molecular players of the autophagy–cell death cross-talk
- •4. AUTOPHAGY, CELLULAR DEATH, AND CANCER
- •5. CONCLUDING REMARKS AND PENDING QUESTIONS
- •SUGGESTED READINGS
- •8 Cell Death in Response to Genotoxic Stress and DNA Damage
- •1. TYPES OF DNA DAMAGE AND REPAIR SYSTEMS
- •2. DNA DAMAGE RESPONSE
- •2.2. Transducers
- •2.3. Effectors
- •4. CHROMATIN MODIFICATIONS
- •5. CELL CYCLE CHECKPOINT REGULATION
- •6. WHEN REPAIR FAILS: SENESCENCE VERSUS APOPTOSIS
- •6.1. DNA damage response and the induction of apoptosis
- •6.2. p53-independent mechanisms of apoptosis
- •6.3. DNA damage response and senescence induction
- •7. DNA DAMAGE FROM OXIDATIVE STRESS
- •SUGGESTED READINGS
- •9 Ceramide and Lipid Mediators in Apoptosis
- •1. INTRODUCTION
- •3.1. Basic cell signaling often involves small molecules
- •3.2. Sphingolipids are cell-signaling molecules
- •3.2.1. Ceramide induces apoptosis
- •3.2.2. Ceramide accumulates during programmed cell death
- •3.2.3. Inhibition of ceramide production alters cell death signaling
- •4.1. Ceramide is generated through SM hydrolysis
- •4.3. aSMase can be activated independently of extracellular receptors to regulate apoptosis
- •4.4. Controversial aspects of the role of aSMase in apoptosis
- •4.5. De novo ceramide synthesis regulates programmed cell death
- •4.6. p53 and Bcl-2–like proteins are connected to de novo ceramide synthesis
- •4.7. The role and regulation of de novo synthesis in ceramide-mediated cell death is poorly understood
- •5. CONCLUDING REMARKS AND FUTURE DIRECTIONS
- •5.1. Who? (Which enzyme?)
- •5.2. What? (Which ceramide?)
- •5.3. Where? (Which compartment?)
- •5.4. When? (At what steps?)
- •5.5. How? (Through what mechanisms?)
- •5.6. What purpose?
- •6. SUMMARY
- •SUGGESTED READINGS
- •1. General Introduction
- •1.1. Cytotoxic lymphocytes and apoptosis
- •2. CYTOTOXIC GRANULES AND GRANULE EXOCYTOSIS
- •2.1. Synthesis and loading of the cytotoxic granule proteins into the secretory granules
- •2.2. The immunological synapse
- •2.3. Secretion of granule proteins
- •2.4. Uptake of proapoptotic proteins into the target cell
- •2.5. Activation of death pathways by granzymes
- •3. GRANULE-BOUND CYTOTOXIC PROTEINS
- •3.1. Perforin
- •3.2. Granulysin
- •3.3. Granzymes
- •3.3.1. GrB-mediated apoptosis
- •3.3.2. GrA-mediated cell death
- •3.3.3. Orphan granzyme-mediated cell death
- •5. CONCLUSIONS
- •REFERENCES
- •Part II Cell Death in Tissues and Organs
- •1.1. Death by trophic factor deprivation
- •1.2. Key molecules regulating neuronal apoptosis during development
- •1.2.1. Roles of caspases and Apaf-1 in neuronal cell death
- •1.2.2. Role of Bcl-2 family members in neuronal cell death
- •1.3. Signal transduction from neurotrophins and neurotrophin receptors
- •1.3.1. Signals for survival
- •1.3.2. Signals for death
- •2.1. Apoptosis in neurodegenerative diseases
- •2.1.4. Amyotrophic lateral sclerosis
- •2.2. Necrotic cell death in neurodegenerative diseases
- •2.2.1. Calpains
- •2.2.2. Cathepsins
- •3. CONCLUSIONS
- •ACKNOWLEDGMENT
- •SUGGESTED READINGS
- •ACKNOWLEDGMENT
- •SUGGESTED READINGS
- •1. INTRODUCTION
- •5. S-NITROSYLATION OF PARKIN
- •7. POTENTIAL TREATMENT OF EXCESSIVE NMDA-INDUCED Ca2+ INFLUX AND FREE RADICAL GENERATION
- •8. FUTURE THERAPEUTICS: NITROMEMANTINES
- •9. CONCLUSIONS
- •Acknowledgments
- •SUGGESTED READINGS
- •3. MITOCHONDRIAL PERMEABILITY TRANSITION ACTIVATED BY Ca2+ AND OXIDATIVE STRESS
- •4.1. Mitochondrial apoptotic pathways
- •4.2. Bcl-2 family proteins
- •4.3. Caspase-dependent apoptosis
- •4.4. Caspase-independent apoptosis
- •4.5. Calpains in ischemic neural cell death
- •5. SUMMARY
- •ACKNOWLEDGMENTS
- •SUGGESTED READINGS
- •1. INTRODUCTION
- •2. HISTORICAL ANTECEDENTS
- •7.1. Activation of p21 waf1/cip1: Targeting extrinsic and intrinsic pathways to death
- •8. CONCLUSION
- •ACKNOWLEDGMENTS
- •REFERENCES
- •16 Apoptosis and Homeostasis in the Eye
- •1.1. Lens
- •1.2. Retina
- •2. ROLE OF APOPTOSIS IN DISEASES OF THE EYE
- •2.1. Glaucoma
- •2.2. Age-related macular degeneration
- •4. APOPTOSIS AND OCULAR IMMUNE PRIVILEGE
- •5. CONCLUSIONS
- •SUGGESTED READINGS
- •17 Cell Death in the Inner Ear
- •3. THE COCHLEA IS THE HEARING ORGAN
- •3.1. Ototoxic hair cell death
- •3.2. Aminoglycoside-induced hair cell death
- •3.3. Cisplatin-induced hair cell death
- •3.4. Therapeutic strategies to prevent hair cell death
- •3.5. Challenges to studies of hair cell death
- •4. SPIRAL GANGLION NEURON DEATH
- •4.1. Neurotrophic support from sensory hair cells and supporting cells
- •4.2. Afferent activity from hair cells
- •4.3. Molecular manifestations of spiral ganglion neuron death
- •4.4. Therapeutic interventions to prevent SGN death
- •ACKNOWLEDGMENTS
- •SUGGESTED READINGS
- •18 Cell Death in the Olfactory System
- •1. Introduction
- •2. Anatomical Aspects
- •3. Life and Death in the Olfactory System
- •3.1. Olfactory epithelium
- •3.2. Olfactory bulb
- •REFERENCES
- •1. Introduction
- •3.1. Beta cell death in the development of T1D
- •3.2. Mechanisms of beta cell death in type 1 diabetes
- •3.2.1. Apoptosis signaling pathways downstream of death receptors and inflammatory cytokines
- •3.2.2. Oxidative stress
- •3.3. Mechanisms of beta cell death in type 2 diabetes
- •3.3.1. Glucolipitoxicity
- •3.3.2. Endoplasmic reticulum stress
- •5. SUMMARY
- •Acknowledgments
- •REFERENCES
- •20 Apoptosis in the Physiology and Diseases of the Respiratory Tract
- •1. APOPTOSIS IN LUNG DEVELOPMENT
- •2. APOPTOSIS IN LUNG PATHOPHYSIOLOGY
- •2.1. Apoptosis in pulmonary inflammation
- •2.2. Apoptosis in acute lung injury
- •2.3. Apoptosis in chronic obstructive pulmonary disease
- •2.4. Apoptosis in interstitial lung diseases
- •2.5. Apoptosis in pulmonary arterial hypertension
- •2.6. Apoptosis in lung cancer
- •SUGGESTED READINGS
- •21 Regulation of Cell Death in the Gastrointestinal Tract
- •1. INTRODUCTION
- •2. ESOPHAGUS
- •3. STOMACH
- •4. SMALL AND LARGE INTESTINE
- •5. LIVER
- •6. PANCREAS
- •7. SUMMARY AND CONCLUDING REMARKS
- •SUGGESTED READINGS
- •22 Apoptosis in the Kidney
- •1. NORMAL KIDNEY STRUCTURE AND FUNCTION
- •3. APOPTOSIS IN ADULT KIDNEY DISEASE
- •4. REGULATION OF APOPTOSIS IN KIDNEY CELLS
- •4.1. Survival factors
- •4.2. Lethal factors
- •4.2.1. TNF superfamily cytokines
- •4.2.2. Other cytokines
- •4.2.3. Glucose
- •4.2.4. Drugs and xenobiotics
- •4.2.5. Ischemia-reperfusion and sepsis
- •5. THERAPEUTIC APPROACHES
- •SUGGESTED READINGS
- •1. INTRODUCTION
- •2. APOPTOSIS IN THE NORMAL BREAST
- •2.1. Occurrence and role of apoptosis in the developing breast
- •2.2.2. Death ligands and death receptor pathway
- •2.2.4. LIF-STAT3 proapoptotic signaling
- •2.2.5. IGF survival signaling
- •2.2.6. Regulation by adhesion
- •2.2.7. PI3K/AKT pathway: molecular hub for survival signals
- •2.2.8. Downstream regulators of apoptosis: the BCL-2 family members
- •3. APOPTOSIS IN BREAST CANCER
- •3.1. Apoptosis in breast tumorigenesis and cancer progression
- •3.2. Molecular dysregulation of apoptosis in breast cancer
- •3.2.1. Altered expression of death ligands and their receptors in breast cancer
- •3.2.2. Deregulation of prosurvival growth factors and their receptors
- •3.2.3. Alterations in cell adhesion and resistance to anoikis
- •3.2.4. Enhanced activation of the PI3K/AKT pathway in breast cancer
- •3.2.5. p53 inactivation in breast cancer
- •3.2.6. Altered expression of BCL-2 family of proteins in breast cancer
- •5. CONCLUSION
- •SUGGESTED READINGS
- •1. INTRODUCTION
- •2. DETECTING CELL DEATH IN THE FEMALE GONADS
- •4. APOPTOSIS AND FEMALE REPRODUCTIVE AGING
- •6. CONCLUDING REMARKS
- •REFERENCES
- •25 Apoptotic Signaling in Male Germ Cells
- •1. INTRODUCTION
- •3.1. Murine models
- •3.2. Primate models
- •3.3. Pathways of caspase activation and apoptosis
- •3.4. Apoptotic signaling in male germ cells
- •5. P38 MITOGEN-ACTIVATED PROTEIN KINASE (MAPK) AND NITRIC OXIDE (NO)–MEDIATED INTRINSIC PATHWAY SIGNALING CONSTITUTES A CRITICAL COMPONENT OF APOPTOTIC SIGNALING IN MALE GERM CELLS AFTER HORMONE DEPRIVATION
- •11. CONCLUSIONS AND PERSPECTIVES
- •REFERENCES
- •26 Cell Death in the Cardiovascular System
- •1. INTRODUCTION
- •2. CELL DEATH IN THE VASCULATURE
- •2.1. Apoptosis in the developing blood vessels
- •2.2. Apoptosis in atherosclerosis
- •2.2.1. Vascular smooth muscle cells
- •2.2.2. Macrophages
- •2.2.3. Regulation of apoptosis in atherosclerosis
- •2.2.4. Necrosis and autophagy in atherosclerosis
- •3. CELL DEATH IN THE MYOCARDIUM
- •3.1. Cell death in myocardial infarction
- •3.1.1. Apoptosis in myocardial infarction
- •3.1.2. Necrosis in myocardial infarction
- •3.1.3. Autophagy in myocardial infarction
- •3.2. Cell death in heart failure
- •3.2.1. Apoptosis in heart failure
- •3.2.2. Necrosis in heart failure
- •3.2.3. Autophagy in heart failure
- •4. CONCLUDING REMARKS
- •ACKNOWLEDGMENTS
- •REFERENCES
- •27 Cell Death Regulation in Muscle
- •1. INTRODUCTION TO MUSCLE
- •1.1. Skeletal muscle adaptation to endurance training
- •1.2. Myonuclear domains
- •2. MITOCHONDRIALLY MEDIATED APOPTOSIS IN MUSCLE
- •2.1. Skeletal muscle apoptotic susceptibility
- •4. APOPTOSIS IN MUSCLE DURING AGING AND DISEASE
- •4.1. Aging
- •4.2. Type 2 diabetes mellitus
- •4.3. Cancer cachexia
- •4.4. Chronic heart failure
- •6. CONCLUSION
- •SUGGESTED READINGS
- •28 Cell Death in the Skin
- •1. INTRODUCTION
- •2. CELL DEATH IN SKIN HOMEOSTASIS
- •2.1. Cornification and apoptosis
- •2.2. Death receptors in the skin
- •3. CELL DEATH IN SKIN PATHOLOGY
- •3.1. Sunburn
- •3.2. Skin cancer
- •3.3. Necrolysis
- •3.4. Pemphigus
- •3.5. Eczema
- •3.6. Graft-versus-host disease
- •4. CONCLUDING REMARKS AND PERSPECTIVES
- •ACKNOWLEDGMENTS
- •SUGGESTED READINGS
- •29 Apoptosis and Cell Survival in the Immune System
- •2.1. Survival of early hematopoietic progenitors
- •2.2. Sizing of the T-cell population
- •2.2.1. Establishing central tolerance
- •2.2.2. Peripheral tolerance
- •2.2.3. Memory T cells
- •2.3. Control of apoptosis in B-cell development
- •2.3.1. Early B-cell development
- •2.3.2. Deletion of autoreactive B cells
- •2.3.3. Survival and death of activated B cells
- •3. IMPAIRED APOPTOSIS AND LEUKEMOGENESIS
- •4. CONCLUSIONS
- •ACKNOWLEDGMENTS
- •REFERENCES
- •30 Cell Death Regulation in the Hematopoietic System
- •1. INTRODUCTION
- •2. HEMATOPOIETIC STEM CELLS
- •4. ERYTHROPOIESIS
- •5. MEGAKARYOPOIESIS
- •6. GRANULOPOIESIS
- •7. MONOPOIESIS
- •8. CONCLUSION
- •ACKNOWLEDGMENTS
- •REFERENCES
- •31 Apoptotic Cell Death in Sepsis
- •1. INTRODUCTION
- •2. HOST INFLAMMATORY RESPONSE TO SEPSIS
- •3. CLINICAL OBSERVATIONS OF CELL DEATH IN SEPSIS
- •3.1. Sepsis-induced apoptosis
- •3.2. Necrotic cell death in sepsis
- •4.1. Central role of apoptosis in sepsis mortality: immune effector cells and gut epithelium
- •4.2. Apoptotic pathways in sepsis-induced immune cell death
- •4.3. Investigations implicating the extrinsic apoptotic pathway in sepsis
- •4.4. Investigations implicating the intrinsic apoptotic pathway in sepsis
- •5. THE EFFECT OF APOPTOSIS ON THE IMMUNE SYSTEM
- •5.1. Cellular effects of an increased apoptotic burdens
- •5.2. Network effects of selective loss of immune cell types
- •5.3. Studies of immunomodulation by apoptotic cells in other fields
- •7. CONCLUSION
- •REFERENCES
- •32 Host–Pathogen Interactions
- •1. INTRODUCTION
- •2. FROM THE PATHOGEN PERSPECTIVE
- •2.1. Commensals versus pathogens
- •2.2. Pathogen strategies to infect the host
- •3. HOST DEFENSE
- •3.1. Antimicrobial peptides
- •3.2. PRRs and inflammation
- •3.2.1. TLRs
- •3.2.2. NLRs
- •3.2.3. The Nod signalosome
- •3.2.4. The inflammasome
- •3.3. Cell death
- •3.3.1. Apoptosis and pathogen clearance
- •3.3.2. Pyroptosis
- •3.2.3. Caspase-independent cell death
- •3.2.4. Autophagy and autophagic cell death
- •4. CONCLUSIONS
- •REFERENCES
- •Part III Cell Death in Nonmammalian Organisms
- •1. PHENOTYPE AND ASSAYS OF YEAST APOPTOSIS
- •2.1. Pheromone-induced cell death
- •2.1.1. Colony growth
- •2.1.2. Killer-induced cell death
- •3. EXTERNAL STIMULI THAT INDUCE APOPTOSIS IN YEAST
- •4. THE GENETICS OF YEAST APOPTOSIS
- •5. PROGRAMMED AND ALTRUISTIC AGING
- •SUGGESTED READINGS
- •34 Caenorhabditis elegans and Apoptosis
- •1. Overview
- •2. KILLING
- •3. SPECIFICATION
- •4. EXECUTION
- •4.1. DNA degradation
- •4.2. Mitochondrial elimination
- •4.3. Engulfment
- •5. SUMMARY
- •SUGGESTED READINGS
- •35 Apoptotic Cell Death in Drosophila
- •2. DROSOPHILA CASPASES AND PROXIMAL REGULATORS
- •6. CLOSING COMMENTS
- •SUGGESTED READINGS
- •36 Analysis of Cell Death in Zebrafish
- •1. INTRODUCTION
- •2. WHY USE ZEBRAFISH TO STUDY CELL DEATH?
- •2.2. Molecular techniques to rapidly assess gene function in embryos
- •2.2.1. Studies of gene function using microinjections into early embryos
- •2.2.2. In situ hybridization and immunohistochemistry
- •2.3. Forward genetic screening
- •2.4. Drug and small-molecule screening
- •2.5. Transgenesis
- •2.6. Targeted knockouts
- •3.1. Intrinsic apoptosis
- •3.2. Extrinsic apoptosis
- •3.3. Chk-1 suppressed apoptosis
- •3.4. Anoikis
- •3.5. Autophagy
- •3.6. Necrosis
- •4. DEVELOPMENTAL CELL DEATH IN ZEBRAFISH EMBRYOS
- •5. THE P53 PATHWAY
- •6. PERSPECTIVES AND FUTURE DIRECTIONS
- •SUGGESTED READING
30 Cell Death Regulation in the Hematopoietic System
Paul A. Ney
1. INTRODUCTION
The hematopoietic system is a dynamic multilineage organ, from which we can gain insights into the physiologic roles of cell death pathways. The role of hematopoiesis is to produce blood cells under normal and stress conditions throughout the lifespan of an organism. In humans, effective function of the hematopoietic system entails the steady and regulated production of erythrocytes, which carry oxygen; platelets, which prevent bleeding; and granulocytes, monocytes, and lymphocytes, which are required for host defense. Remarkably, these varied cell types are derived from a common ancestor, the hematopoietic stem cell (HSC). HSCs are known for certain key properties: they are long-lived and self-renewing and give rise to a steady supply of multipotential progenitor cells (MPPs) that are committed to undergo differentiation. MPPs in turn give rise to progenitors with a progressively restricted developmental potential; these undergo regulated expansion and, eventually, commitment to terminal differentiation. The terminal differentiation program is characterized by decreased proliferation or mitotic arrest, global changes in gene expression, and cellular remodeling. Finally, cells that have fulfilled their purpose, or senescent cells, are cleared from the body to make room for new cells, or after a stress response to reestablish homeostasis.
As discussed in other chapters, cell death pathways can be divided, on the basis of their mechanism of action, into extrinsic and intrinsic arms. In a simplified view, extrinsic death pathways are activated by the binding of extracellular ligands to members of the death receptor family. These include Fas, tumor necrosis factor (TNF) family receptors, and TNF-related apoptosisinducing ligand (TRAIL) family receptors.1,2,3 Activation
of death receptors leads to the assembly of a deathinducing signaling complex, which in turn leads to activation of apical caspases (caspase-8 in mice, caspase-8 and -10 in humans) and cleavage and subsequent activation of effector caspases (caspase-3 and -7). Activation of caspase-3 and -7 leads to cleavage of caspase targets, which include regulatory, structural, and homeostatic proteins, and to programmed cell death.4 Intrinsic death pathways are activated by cellular stress, such as DNA damage or growth factor deprivation, and these signal to mitochondria through the BCL2 family of proteins, causing cytochrome c release, assembly of the apoptosome, and activation of effector caspases.5
The BCL2 family of proteins can be divided into antiapoptotic and proapoptotic groups.6 Antiapoptotic BCL2-related proteins typically contain four BCL2homology domains (BH1-BH4); these include BCL2, BCL-XL, A1, BCL-W, and BOO. Proapoptotic BCL2related proteins can be further subdivided into a multidomain proapoptotic group – BAX, BAK, and BOK – which contains three BCL2-homology domains (BH1BH3), and a second larger group, which contains a single BCL2-homology domain (BH3-only proteins). In response to cellular stress, BH3-only proteins transduce proapoptotic signals to mitochondria where they directly or indirectly activate BAX or BAK,7,8 causing cytochrome c release and apoptosis.9,10 Additionally, a BH3-only protein, BID, can be cleaved and activated by caspase-8, linking the extrinsic and intrinsic pathways.11,12
Distinct phases of hematopoietic development are under the control of cell death pathways; these include survival of stem cells, expansion and lineage determination of progenitor cells, terminal differentiation of precursor cells, function of formed blood cells, and the elimination of senescent cells. Therefore, the purposes of this
350

CELL DEATH REGULATION IN THE HEMATOPOIETIC SYSTEM |
351 |
Apoptosis |
HSC |
MPP |
Figure 30-1. Hematopoietic stem cell fates. HSCs can either undergo self-renewal, di erentiation to an MPP, or apoptosis. Depending on whether cell divisions are symmetric (give rise to identical progeny) or asymmetric (give rise to progeny with di erent fates), HSCs may increase in numbers, decrease, or stay the same.
chapter are (1) to review the roles of the extrinsic and intrinsic cell death pathways in hematopoietic development at each of these stages; and (2) to place these findings in the broader biological context of the regulation of cellular proliferation and differentiation.
2. HEMATOPOIETIC STEM CELLS
Long-term repopulating HSCs provide a steady supply of multipotential progenitor cells throughout the lifespan of an organism. Depletion of HSC leads to hematopoietic insufficiency and death. Therefore, HSC must be protected and the HSC pool maintained over an extended period of time. There are several potential cellular fates of HSC; these include self-renewal, proliferation, differentiation, and programmed cell death (Figure 30-1). These fates must be balanced to sustain a functioning HSC pool throughout the lifespan of an organism.
Chief among the mechanisms that preserve HSCs is their quiescent cell cycle status; the majority of HSCs are in the G0 phase of the cell cycle and undergo cell division about once per month.13 Infrequent cell division protects HSCs from cell cycle checkpoint-activated apoptosis; indeed, HSCs accumulate DNA damage over time, but their reserves are not significantly depleted, and a clear deficit in stem cell function only emerges under stress conditions, such as transplantation, when HSC are induced to divide.14,15,16 Another key mechanism for maintenance of the HSC pool is repression of differentiation potential. The HSC transcriptome overlaps that of other types of stem cells and includes genes that are important for self-renewal.17,18 When HSCs commit to differentiation, these genes are downregulated, and in a process known as multilineage priming, other genes characteristic of multiple hematopoietic lineages are expressed.19 Certain transcription factors and corepressors inhibit this fate change, and deficiencies of these factors are characterized by stem-cell depletion and other hematopoietic abnormalities.20,21,22,23
The bone marrow microenvironment or niche is important for maintaining HSCs in a quiescent state and promoting HSC survival.24 Stromal cells in the bone marrow microenvironment produce cytokines, which are implicated in HSC homeostasis; these include stemcell factor, thrombopoietin, notch ligands, Wnts, and angiopoietin-1.25 Among these, stem-cell factor, and additionally the growth factor Flt3 ligand, upregulate MCL1 in HSCs.26,27 MCL1 is highly expressed in HSCs and is essential for HSC survival.26 On the other hand, BCL2 and BCL-XL are dispensable.28,29 Overall, the role of apoptosis in HSC homeostasis was addressed through the use of Tg(H2k-Bcl2) mice, in which BCL2 expression is enforced by a constitutive promoter.30 HSCs from these mice are expanded 2.4-fold as compared with wildtype controls and can out-compete wild-type HSCs in transplantation experiments. Thus inhibition of apoptosis is critical for HSC survival, but apoptosis has a limited role in the regulation of HSC numbers.
For the most part, the signal transduction pathways that regulate HSC homeostasis remain to be defined. One exception is Akt, which appears to play an essential role. Mutation of Pten, a negative regulator of Akt, leads to increased HSC cycling and depletion.31 Furthermore, the FoxO transcription factor family, which is negatively regulated by Akt, has a role in the maintenance of HSC quiescence through its inhibitory effect on reactive oxygen species (ROS) generation.32
3. HEMATOPOIETIC PROGENITOR EXPANSION AND
LINEAGE DETERMINATION
Regulation of HSC commitment to differentiation is not well understood, but may be related to disengagement of HSC from their specialized niche.24 Once HSCs commit to differentiate, they start to cycle more frequently, lose their capacity for self-renewal, and develop into MPPs. The role of MPPs is to supply the proper number of differentiated hematopoietic cells to meet the immediate needs of the organism. To accomplish this, two processes are concurrently engaged and subject to feedback regulation: the first is regulation of cell numbers, and the second is lineage determination. With respect to the former, calculations of potential versus actual hematopoietic cell production show that a high percentage of early hematopoietic progenitors undergo cell death.33 Thus MPPs and their progeny provide a significant reservoir of hematopoietic production.
Similar to HSC, MCL1 expression in hematopoietic progenitors is regulated by stem-cell factor, and MCL1 is essential for progenitor cell survival.26 However, although Tg(Vav-Bcl2), Bim–/–, and Bax–/–; Bak–/– mice
352 |
PAUL A. NEY |
all exhibit hematopoietic expansion,34,35,36 the effect on progenitors is minimal, suggesting that downregulation of the intrinsic proapoptotic pathway primarily affects the survival of post-progenitor cells. In contrast, lpr/lpr and gld/gld mice, which are defective for FASFASL signaling, exhibit marked extramedullary expansion of hematopoietic progenitors,37 suggesting that the extrinsic death pathway has an important role in the regulation of progenitor expansion.
With regard to lineage determination, the underlying mechanism is unresolved. It is apparent that once HSCs commit to differentiation, a cell-intrinsic program is activated that inexorably leads to terminal differentiation or cell death. General features of this program include passage through a series of lineage-determining branch points,38,39 a progressive reduction in transcriptome complexity,40,41 and shortening of the length of the cell cycle.33 There are two main theories on the mechanism of lineage determination. One is the random or stochastic model. The ability of BCL2 to rescue lineagespecific hematopoietic development in mice lacking functional M-CSF or IL-7Rα supports the idea that lineage is determined by random cell-intrinsic events, and cytokines play a permissive role in the expansion of specific lineages through their effect on cell survival.42,43 A second is the instructive model. In support of this model, it has been shown that cytokine signaling has the ability to reprogram lineage-restricted progenitors.44 Finally, a hybrid model is suggested wherein bipotential progenitors exist in a metastable state, reinforced by positive feedback loops, and both random and deterministic events play a role in lineage specification.45 An attractive aspect of this model is that it provides a theoretical basis for the existence of multipotential progenitor pools.
In summary, hematopoiesis at the progenitor stage is regulated in a manner to allow the rapid, but self-limited, expansion of specific lineages in response to peripheral signals from the organism. In the remainder of this chapter, variations on this theme are discussed for each of the major lineages, as well as the role of cell death pathways in the formation and function of mature blood cells. One exception is lymphocytes, which are discussed elsewhere in this volume.
4. ERYTHROPOIESIS
The bipotential megakaryocyte-erythroid (MEP) progenitor gives rise to cells that are committed to the megakaryocytic or erythroid lineages. In the erythroid lineage, the burst-forming unit-erythroid (BFU-E) is the first committed progenitor to develop, and the colonyforming unit-erythroid (CFU-E) is the second. BFU-E
SCF,
|
|
|
|
BFU-E |
IL-3 |
|
|||
|
|
|||
|
|
|
|
CFU-E |
|
|
|
|
|
|
|
|
|
|
|
|
|
||
Pro-E
marrow Bone
Baso-E
|
|
FAS- |
|
|
FASL |
|
Ortho-E |
BCL-XL |
|
Retic |
NIX |
|
Erythrocyte |
Blood |
|
|
|
EPO |
delivery |
Tissue |
|
oxygen |
|
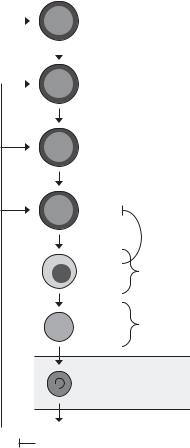
Figure 30-2. Regulation of erythropoiesis by erythropoietin (EPO). EPO is required for erythroid cell survival beginning at approximately the proerythroblast (Pro-E) stage; further, under conditions of erythropoietic stress, EPO also causes expansion of CFU-E. FASLexpressing orthochromatic erythroblasts (Ortho-E) are postulated to induce apoptosis of FAS-expressing basophilic erythroblasts (Baso-E), which is opposed by EPO. NIX and BCL-XL are upregulated during the terminal, EPO-independent, phase of di erentiation. IL-3, interleukin- 3; Retic, reticulocytes; SCF, stem cell factor.
have greater proliferative potential than CFU-E, and give rise to colonies that contain hundreds to thousands of erythroid cells. CFU-E are more mature, and give rise to colonies of 8–64 erythroid cells. Erythroid development beyond the CFU-E stage follows a stereotypical program, consisting of a limited number of cell divisions (3–6 divisions), mitotic arrest, and terminal differentiation.
The primary pathway involved in the regulation of erythrocyte production is erythropoietin receptor (EPOR) signaling (Figure 30-2). The EPOR is expressed throughout erythroid development, peaks at the CFU- E stage, and declines thereafter.46,47 The EPOR is essential for erythroid cell survival; accordingly, Epor–/– mice die of severe anemia around embryonic day 12.5 (E12.5), after the onset of definitive erythropoiesis in the fetal liver.48 Epor–/– fetal livers contain near normal
CELL DEATH REGULATION IN THE HEMATOPOIETIC SYSTEM |
353 |
numbers of early and late erythroid progenitors, but are blocked at the post–CFU-E stage as a result of apoptosis. The EPOR is a member of the cytokine receptor family and lacks intrinsic tyrosine kinase activity; however, the EPOR membrane-proximal cytoplasmic domain is associated with janus kinase 2 (JAK2).49 Jak2–/– mice also die of severe anemia around E12.5 because of a severe defect of erythroid cell survival, demonstrating that JAK2 is essential for EPOR function.50 BCL-XL is highly expressed in erythroid cells,51 and it is suggested that BCL-XL is a major downstream target of EPOR signaling52,53; indeed, BCL-XL is essential for erythroid cell survival, and enforced expression of BCL-XL can supplant the requirement for erythropoietin (EPO) at the final stages of erythroid differentiation.29,54 On the other hand, EPO supports the survival of BCL-XL–deficient basophilic erythroblasts, and the death of BCL-XL–deficient erythroblasts occurs at a late stage of maturation.55,56 Thus BCL- XL appears not to be the primary target of EPOR and JAK2 signaling that supports erythroid cell survival.
EPO deprivation causes caspase activation and apoptosis.51,57,58 The fact that BCL-XL can rescue the survival of EPO-deprived erythroid cells suggests that death in this setting is mediated at least in part through the intrinsic pathway.54 On the other hand, mature erythroblasts express membrane-bound FASL, and immature erythroblasts are sensitive to FAS-mediated apoptosis through the extrinsic pathway.1,59 Because these cells are in direct contact in three-dimensional erythroblastic islands in vivo,60 it has been suggested that erythropoiesis is negatively autoregulated (Figure 30-2).59,61 The pattern of TRAIL and TRAIL receptor expression is also consistent with this model.62 Results from mutant mouse strains, however, are inconclusive: FAS-FASL signaling-deficient lpr/lpr and gld/gld mice do not have an overt erythroid phenotype, but Caspase8–/– and Fadd–/– mice exhibit erythrocytosis.63,64 Thus additional studies are needed to elucidate the death pathway opposed by EPOR signaling.
From the CFU-E stage onward, erythroblasts undergo a series of three to six terminal cell divisions and a morphological transformation. This stereotypical program is characterized by a decrease in cell size,65 mitotic arrest,66 nuclear condensation and expulsion, global changes in gene expression and mRNA splicing,67,68 and a dramatic increase in globin gene expression.69 Caspase-3 and -7 are transiently activated1,70 and may have a role in erythroid maturation. In this regard, either siRNA knockdown of caspase-3 or caspase inhibitors cause a differentiation block at the proerythroblast stage, but caspase inhibitors have no effect at later stages of erythroid differentiation.70,71 Consistent with the latter, there is no
relationship between cleavage or degradation of nuclear subcompartment proteins and developmentally regulated nuclear condensation.72
Nascent mammalian reticulocytes generated by the enucleation of mature erythroblasts reside in the bone marrow for 24 hours, are released into circulation, and complete differentiation over 2 to 3 days to form mature erythrocytes.73,74 During this time, reticulocytes change from large, motile, multilobular cells to small, biconcave, discoid cells75; lose surface area and volume76; undergo cytoskeletal and membrane remodeling77; and eliminate ribosomes and membrane-bound organelles.78,79 Autophagy, or programmed self-digestion, is the process of envelopment of cytoplasm and organelles by doublemembraned vesicles and delivery to lysosomes.80 Ultrastructural studies suggest that during reticulocyte maturation, mitochondria are cleared by an autophagyrelated process.78,81 Further, recent studies show that programmed mitochondrial clearance in reticulocytes depends on the autophagy protein, ULK1,82 and the proapoptotic BH3-only protein, NIX.83,84
BCL-XL and NIX are coordinately upregulated during terminal erythroid differentiation (Figure 30-2),83,85 and it is suggested that NIX may regulate erythrocyte production by providing a proapoptotic signal that opposes BCL-XL.86 However, contrary to this notion, the erythroid defect caused by BCL-XL deficiency is not rescued by loss of NIX (unpublished results). Another possibility, consistent with the role of NIX in mitochondrial clearance, is that upregulation of NIX and BCL-XL may simultaneously serve to promote cellular remodeling and to protect late erythroid cells from apoptosis. In this regard, there are at least three potential mechanisms through which NIX may function. First, in keeping with the well-established function of other BH3-only proteins, NIX may cause mitochondrial depolarization, which in turn causes mitochondrial autophagy.84 However, if this is the mechanism, then it must occur independent of BAX, BAK, and the mitochondrial permeability transition pore, because none of these is required for mitochondrial clearance.83 Second, NIX may function as an adaptor protein to recruit components of the autophagy or membrane trafficking machinery to mitochondria. Third, by analogy to other BH3-only proteins,87 NIX may activate autophagy by competing with the autophagy protein Beclin-1 for binding to BCL-XL. One of these mechanisms may be employed as erythroid cells switch from an apoptosis-prone to an autophagyprone state during terminal differentiation.
Mature human erythrocytes in circulation have a lifespan of approximately 120 days.1 Erythrocyte changes associated with senescence include membrane