

- •Contents
- •Contributors
- •Part I General Principles of Cell Death
- •1 Human Caspases – Apoptosis and Inflammation Signaling Proteases
- •1.1. Apoptosis and limited proteolysis
- •1.2. Caspase evolution
- •2. ACTIVATION MECHANISMS
- •2.2. The activation platforms
- •2.4. Proteolytic maturation
- •3. CASPASE SUBSTRATES
- •4. REGULATION BY NATURAL INHIBITORS
- •REFERENCES
- •2 Inhibitor of Apoptosis Proteins
- •2. CELLULAR FUNCTIONS AND PHENOTYPES OF IAP
- •3. IN VIVO FUNCTIONS OF IAP FAMILY PROTEINS
- •4. SUBCELLULAR LOCATIONS OF IAP
- •8. IAP–IAP INTERACTIONS
- •10. ENDOGENOUS ANTAGONISTS OF IAP
- •11. IAPs AND DISEASE
- •SUGGESTED READINGS
- •1. INTRODUCTION
- •2.1. The CD95 (Fas/APO-1) system
- •2.1.1. CD95 and CD95L: discovery of the first direct apoptosis-inducing receptor-ligand system
- •2.1.2. Biochemistry of CD95 apoptosis signaling
- •2.2. The TRAIL (Apo2L) system
- •3.1. The TNF system
- •3.1.1. Biochemistry of TNF signal transduction
- •3.1.2. TNF and TNF blockers in the clinic
- •3.2. The DR3 system
- •4. THE DR6 SYSTEM
- •6. CONCLUDING REMARKS AND OUTLOOK
- •SUGGESTED READINGS
- •4 Mitochondria and Cell Death
- •1. INTRODUCTION
- •2. MITOCHONDRIAL PHYSIOLOGY
- •3. THE MITOCHONDRIAL PATHWAY OF APOPTOSIS
- •9. CONCLUSIONS
- •SUGGESTED READINGS
- •1. INTRODUCTION
- •3. INHIBITING APOPTOSIS
- •4. INHIBITING THE INHIBITORS
- •6. THE BCL-2 FAMILY AND CANCER
- •SUGGESTED READINGS
- •6 Endoplasmic Reticulum Stress Response in Cell Death and Cell Survival
- •1. INTRODUCTION
- •2. THE ESR IN YEAST
- •3. THE ESR IN MAMMALS
- •4. THE ESR AND CELL DEATH
- •5. THE ESR IN DEVELOPMENT AND TISSUE HOMEOSTASIS
- •6. THE ESR IN HUMAN DISEASE
- •7. CONCLUSION
- •7 Autophagy – The Liaison between the Lysosomal System and Cell Death
- •1. INTRODUCTION
- •2. AUTOPHAGY
- •2.2. Physiologic functions of autophagy
- •2.3. Autophagy and human pathology
- •3. AUTOPHAGY AND CELL DEATH
- •3.1. Autophagy as anti–cell death mechanism
- •3.2. Autophagy as a cell death mechanism
- •3.3. Molecular players of the autophagy–cell death cross-talk
- •4. AUTOPHAGY, CELLULAR DEATH, AND CANCER
- •5. CONCLUDING REMARKS AND PENDING QUESTIONS
- •SUGGESTED READINGS
- •8 Cell Death in Response to Genotoxic Stress and DNA Damage
- •1. TYPES OF DNA DAMAGE AND REPAIR SYSTEMS
- •2. DNA DAMAGE RESPONSE
- •2.2. Transducers
- •2.3. Effectors
- •4. CHROMATIN MODIFICATIONS
- •5. CELL CYCLE CHECKPOINT REGULATION
- •6. WHEN REPAIR FAILS: SENESCENCE VERSUS APOPTOSIS
- •6.1. DNA damage response and the induction of apoptosis
- •6.2. p53-independent mechanisms of apoptosis
- •6.3. DNA damage response and senescence induction
- •7. DNA DAMAGE FROM OXIDATIVE STRESS
- •SUGGESTED READINGS
- •9 Ceramide and Lipid Mediators in Apoptosis
- •1. INTRODUCTION
- •3.1. Basic cell signaling often involves small molecules
- •3.2. Sphingolipids are cell-signaling molecules
- •3.2.1. Ceramide induces apoptosis
- •3.2.2. Ceramide accumulates during programmed cell death
- •3.2.3. Inhibition of ceramide production alters cell death signaling
- •4.1. Ceramide is generated through SM hydrolysis
- •4.3. aSMase can be activated independently of extracellular receptors to regulate apoptosis
- •4.4. Controversial aspects of the role of aSMase in apoptosis
- •4.5. De novo ceramide synthesis regulates programmed cell death
- •4.6. p53 and Bcl-2–like proteins are connected to de novo ceramide synthesis
- •4.7. The role and regulation of de novo synthesis in ceramide-mediated cell death is poorly understood
- •5. CONCLUDING REMARKS AND FUTURE DIRECTIONS
- •5.1. Who? (Which enzyme?)
- •5.2. What? (Which ceramide?)
- •5.3. Where? (Which compartment?)
- •5.4. When? (At what steps?)
- •5.5. How? (Through what mechanisms?)
- •5.6. What purpose?
- •6. SUMMARY
- •SUGGESTED READINGS
- •1. General Introduction
- •1.1. Cytotoxic lymphocytes and apoptosis
- •2. CYTOTOXIC GRANULES AND GRANULE EXOCYTOSIS
- •2.1. Synthesis and loading of the cytotoxic granule proteins into the secretory granules
- •2.2. The immunological synapse
- •2.3. Secretion of granule proteins
- •2.4. Uptake of proapoptotic proteins into the target cell
- •2.5. Activation of death pathways by granzymes
- •3. GRANULE-BOUND CYTOTOXIC PROTEINS
- •3.1. Perforin
- •3.2. Granulysin
- •3.3. Granzymes
- •3.3.1. GrB-mediated apoptosis
- •3.3.2. GrA-mediated cell death
- •3.3.3. Orphan granzyme-mediated cell death
- •5. CONCLUSIONS
- •REFERENCES
- •Part II Cell Death in Tissues and Organs
- •1.1. Death by trophic factor deprivation
- •1.2. Key molecules regulating neuronal apoptosis during development
- •1.2.1. Roles of caspases and Apaf-1 in neuronal cell death
- •1.2.2. Role of Bcl-2 family members in neuronal cell death
- •1.3. Signal transduction from neurotrophins and neurotrophin receptors
- •1.3.1. Signals for survival
- •1.3.2. Signals for death
- •2.1. Apoptosis in neurodegenerative diseases
- •2.1.4. Amyotrophic lateral sclerosis
- •2.2. Necrotic cell death in neurodegenerative diseases
- •2.2.1. Calpains
- •2.2.2. Cathepsins
- •3. CONCLUSIONS
- •ACKNOWLEDGMENT
- •SUGGESTED READINGS
- •ACKNOWLEDGMENT
- •SUGGESTED READINGS
- •1. INTRODUCTION
- •5. S-NITROSYLATION OF PARKIN
- •7. POTENTIAL TREATMENT OF EXCESSIVE NMDA-INDUCED Ca2+ INFLUX AND FREE RADICAL GENERATION
- •8. FUTURE THERAPEUTICS: NITROMEMANTINES
- •9. CONCLUSIONS
- •Acknowledgments
- •SUGGESTED READINGS
- •3. MITOCHONDRIAL PERMEABILITY TRANSITION ACTIVATED BY Ca2+ AND OXIDATIVE STRESS
- •4.1. Mitochondrial apoptotic pathways
- •4.2. Bcl-2 family proteins
- •4.3. Caspase-dependent apoptosis
- •4.4. Caspase-independent apoptosis
- •4.5. Calpains in ischemic neural cell death
- •5. SUMMARY
- •ACKNOWLEDGMENTS
- •SUGGESTED READINGS
- •1. INTRODUCTION
- •2. HISTORICAL ANTECEDENTS
- •7.1. Activation of p21 waf1/cip1: Targeting extrinsic and intrinsic pathways to death
- •8. CONCLUSION
- •ACKNOWLEDGMENTS
- •REFERENCES
- •16 Apoptosis and Homeostasis in the Eye
- •1.1. Lens
- •1.2. Retina
- •2. ROLE OF APOPTOSIS IN DISEASES OF THE EYE
- •2.1. Glaucoma
- •2.2. Age-related macular degeneration
- •4. APOPTOSIS AND OCULAR IMMUNE PRIVILEGE
- •5. CONCLUSIONS
- •SUGGESTED READINGS
- •17 Cell Death in the Inner Ear
- •3. THE COCHLEA IS THE HEARING ORGAN
- •3.1. Ototoxic hair cell death
- •3.2. Aminoglycoside-induced hair cell death
- •3.3. Cisplatin-induced hair cell death
- •3.4. Therapeutic strategies to prevent hair cell death
- •3.5. Challenges to studies of hair cell death
- •4. SPIRAL GANGLION NEURON DEATH
- •4.1. Neurotrophic support from sensory hair cells and supporting cells
- •4.2. Afferent activity from hair cells
- •4.3. Molecular manifestations of spiral ganglion neuron death
- •4.4. Therapeutic interventions to prevent SGN death
- •ACKNOWLEDGMENTS
- •SUGGESTED READINGS
- •18 Cell Death in the Olfactory System
- •1. Introduction
- •2. Anatomical Aspects
- •3. Life and Death in the Olfactory System
- •3.1. Olfactory epithelium
- •3.2. Olfactory bulb
- •REFERENCES
- •1. Introduction
- •3.1. Beta cell death in the development of T1D
- •3.2. Mechanisms of beta cell death in type 1 diabetes
- •3.2.1. Apoptosis signaling pathways downstream of death receptors and inflammatory cytokines
- •3.2.2. Oxidative stress
- •3.3. Mechanisms of beta cell death in type 2 diabetes
- •3.3.1. Glucolipitoxicity
- •3.3.2. Endoplasmic reticulum stress
- •5. SUMMARY
- •Acknowledgments
- •REFERENCES
- •20 Apoptosis in the Physiology and Diseases of the Respiratory Tract
- •1. APOPTOSIS IN LUNG DEVELOPMENT
- •2. APOPTOSIS IN LUNG PATHOPHYSIOLOGY
- •2.1. Apoptosis in pulmonary inflammation
- •2.2. Apoptosis in acute lung injury
- •2.3. Apoptosis in chronic obstructive pulmonary disease
- •2.4. Apoptosis in interstitial lung diseases
- •2.5. Apoptosis in pulmonary arterial hypertension
- •2.6. Apoptosis in lung cancer
- •SUGGESTED READINGS
- •21 Regulation of Cell Death in the Gastrointestinal Tract
- •1. INTRODUCTION
- •2. ESOPHAGUS
- •3. STOMACH
- •4. SMALL AND LARGE INTESTINE
- •5. LIVER
- •6. PANCREAS
- •7. SUMMARY AND CONCLUDING REMARKS
- •SUGGESTED READINGS
- •22 Apoptosis in the Kidney
- •1. NORMAL KIDNEY STRUCTURE AND FUNCTION
- •3. APOPTOSIS IN ADULT KIDNEY DISEASE
- •4. REGULATION OF APOPTOSIS IN KIDNEY CELLS
- •4.1. Survival factors
- •4.2. Lethal factors
- •4.2.1. TNF superfamily cytokines
- •4.2.2. Other cytokines
- •4.2.3. Glucose
- •4.2.4. Drugs and xenobiotics
- •4.2.5. Ischemia-reperfusion and sepsis
- •5. THERAPEUTIC APPROACHES
- •SUGGESTED READINGS
- •1. INTRODUCTION
- •2. APOPTOSIS IN THE NORMAL BREAST
- •2.1. Occurrence and role of apoptosis in the developing breast
- •2.2.2. Death ligands and death receptor pathway
- •2.2.4. LIF-STAT3 proapoptotic signaling
- •2.2.5. IGF survival signaling
- •2.2.6. Regulation by adhesion
- •2.2.7. PI3K/AKT pathway: molecular hub for survival signals
- •2.2.8. Downstream regulators of apoptosis: the BCL-2 family members
- •3. APOPTOSIS IN BREAST CANCER
- •3.1. Apoptosis in breast tumorigenesis and cancer progression
- •3.2. Molecular dysregulation of apoptosis in breast cancer
- •3.2.1. Altered expression of death ligands and their receptors in breast cancer
- •3.2.2. Deregulation of prosurvival growth factors and their receptors
- •3.2.3. Alterations in cell adhesion and resistance to anoikis
- •3.2.4. Enhanced activation of the PI3K/AKT pathway in breast cancer
- •3.2.5. p53 inactivation in breast cancer
- •3.2.6. Altered expression of BCL-2 family of proteins in breast cancer
- •5. CONCLUSION
- •SUGGESTED READINGS
- •1. INTRODUCTION
- •2. DETECTING CELL DEATH IN THE FEMALE GONADS
- •4. APOPTOSIS AND FEMALE REPRODUCTIVE AGING
- •6. CONCLUDING REMARKS
- •REFERENCES
- •25 Apoptotic Signaling in Male Germ Cells
- •1. INTRODUCTION
- •3.1. Murine models
- •3.2. Primate models
- •3.3. Pathways of caspase activation and apoptosis
- •3.4. Apoptotic signaling in male germ cells
- •5. P38 MITOGEN-ACTIVATED PROTEIN KINASE (MAPK) AND NITRIC OXIDE (NO)–MEDIATED INTRINSIC PATHWAY SIGNALING CONSTITUTES A CRITICAL COMPONENT OF APOPTOTIC SIGNALING IN MALE GERM CELLS AFTER HORMONE DEPRIVATION
- •11. CONCLUSIONS AND PERSPECTIVES
- •REFERENCES
- •26 Cell Death in the Cardiovascular System
- •1. INTRODUCTION
- •2. CELL DEATH IN THE VASCULATURE
- •2.1. Apoptosis in the developing blood vessels
- •2.2. Apoptosis in atherosclerosis
- •2.2.1. Vascular smooth muscle cells
- •2.2.2. Macrophages
- •2.2.3. Regulation of apoptosis in atherosclerosis
- •2.2.4. Necrosis and autophagy in atherosclerosis
- •3. CELL DEATH IN THE MYOCARDIUM
- •3.1. Cell death in myocardial infarction
- •3.1.1. Apoptosis in myocardial infarction
- •3.1.2. Necrosis in myocardial infarction
- •3.1.3. Autophagy in myocardial infarction
- •3.2. Cell death in heart failure
- •3.2.1. Apoptosis in heart failure
- •3.2.2. Necrosis in heart failure
- •3.2.3. Autophagy in heart failure
- •4. CONCLUDING REMARKS
- •ACKNOWLEDGMENTS
- •REFERENCES
- •27 Cell Death Regulation in Muscle
- •1. INTRODUCTION TO MUSCLE
- •1.1. Skeletal muscle adaptation to endurance training
- •1.2. Myonuclear domains
- •2. MITOCHONDRIALLY MEDIATED APOPTOSIS IN MUSCLE
- •2.1. Skeletal muscle apoptotic susceptibility
- •4. APOPTOSIS IN MUSCLE DURING AGING AND DISEASE
- •4.1. Aging
- •4.2. Type 2 diabetes mellitus
- •4.3. Cancer cachexia
- •4.4. Chronic heart failure
- •6. CONCLUSION
- •SUGGESTED READINGS
- •28 Cell Death in the Skin
- •1. INTRODUCTION
- •2. CELL DEATH IN SKIN HOMEOSTASIS
- •2.1. Cornification and apoptosis
- •2.2. Death receptors in the skin
- •3. CELL DEATH IN SKIN PATHOLOGY
- •3.1. Sunburn
- •3.2. Skin cancer
- •3.3. Necrolysis
- •3.4. Pemphigus
- •3.5. Eczema
- •3.6. Graft-versus-host disease
- •4. CONCLUDING REMARKS AND PERSPECTIVES
- •ACKNOWLEDGMENTS
- •SUGGESTED READINGS
- •29 Apoptosis and Cell Survival in the Immune System
- •2.1. Survival of early hematopoietic progenitors
- •2.2. Sizing of the T-cell population
- •2.2.1. Establishing central tolerance
- •2.2.2. Peripheral tolerance
- •2.2.3. Memory T cells
- •2.3. Control of apoptosis in B-cell development
- •2.3.1. Early B-cell development
- •2.3.2. Deletion of autoreactive B cells
- •2.3.3. Survival and death of activated B cells
- •3. IMPAIRED APOPTOSIS AND LEUKEMOGENESIS
- •4. CONCLUSIONS
- •ACKNOWLEDGMENTS
- •REFERENCES
- •30 Cell Death Regulation in the Hematopoietic System
- •1. INTRODUCTION
- •2. HEMATOPOIETIC STEM CELLS
- •4. ERYTHROPOIESIS
- •5. MEGAKARYOPOIESIS
- •6. GRANULOPOIESIS
- •7. MONOPOIESIS
- •8. CONCLUSION
- •ACKNOWLEDGMENTS
- •REFERENCES
- •31 Apoptotic Cell Death in Sepsis
- •1. INTRODUCTION
- •2. HOST INFLAMMATORY RESPONSE TO SEPSIS
- •3. CLINICAL OBSERVATIONS OF CELL DEATH IN SEPSIS
- •3.1. Sepsis-induced apoptosis
- •3.2. Necrotic cell death in sepsis
- •4.1. Central role of apoptosis in sepsis mortality: immune effector cells and gut epithelium
- •4.2. Apoptotic pathways in sepsis-induced immune cell death
- •4.3. Investigations implicating the extrinsic apoptotic pathway in sepsis
- •4.4. Investigations implicating the intrinsic apoptotic pathway in sepsis
- •5. THE EFFECT OF APOPTOSIS ON THE IMMUNE SYSTEM
- •5.1. Cellular effects of an increased apoptotic burdens
- •5.2. Network effects of selective loss of immune cell types
- •5.3. Studies of immunomodulation by apoptotic cells in other fields
- •7. CONCLUSION
- •REFERENCES
- •32 Host–Pathogen Interactions
- •1. INTRODUCTION
- •2. FROM THE PATHOGEN PERSPECTIVE
- •2.1. Commensals versus pathogens
- •2.2. Pathogen strategies to infect the host
- •3. HOST DEFENSE
- •3.1. Antimicrobial peptides
- •3.2. PRRs and inflammation
- •3.2.1. TLRs
- •3.2.2. NLRs
- •3.2.3. The Nod signalosome
- •3.2.4. The inflammasome
- •3.3. Cell death
- •3.3.1. Apoptosis and pathogen clearance
- •3.3.2. Pyroptosis
- •3.2.3. Caspase-independent cell death
- •3.2.4. Autophagy and autophagic cell death
- •4. CONCLUSIONS
- •REFERENCES
- •Part III Cell Death in Nonmammalian Organisms
- •1. PHENOTYPE AND ASSAYS OF YEAST APOPTOSIS
- •2.1. Pheromone-induced cell death
- •2.1.1. Colony growth
- •2.1.2. Killer-induced cell death
- •3. EXTERNAL STIMULI THAT INDUCE APOPTOSIS IN YEAST
- •4. THE GENETICS OF YEAST APOPTOSIS
- •5. PROGRAMMED AND ALTRUISTIC AGING
- •SUGGESTED READINGS
- •34 Caenorhabditis elegans and Apoptosis
- •1. Overview
- •2. KILLING
- •3. SPECIFICATION
- •4. EXECUTION
- •4.1. DNA degradation
- •4.2. Mitochondrial elimination
- •4.3. Engulfment
- •5. SUMMARY
- •SUGGESTED READINGS
- •35 Apoptotic Cell Death in Drosophila
- •2. DROSOPHILA CASPASES AND PROXIMAL REGULATORS
- •6. CLOSING COMMENTS
- •SUGGESTED READINGS
- •36 Analysis of Cell Death in Zebrafish
- •1. INTRODUCTION
- •2. WHY USE ZEBRAFISH TO STUDY CELL DEATH?
- •2.2. Molecular techniques to rapidly assess gene function in embryos
- •2.2.1. Studies of gene function using microinjections into early embryos
- •2.2.2. In situ hybridization and immunohistochemistry
- •2.3. Forward genetic screening
- •2.4. Drug and small-molecule screening
- •2.5. Transgenesis
- •2.6. Targeted knockouts
- •3.1. Intrinsic apoptosis
- •3.2. Extrinsic apoptosis
- •3.3. Chk-1 suppressed apoptosis
- •3.4. Anoikis
- •3.5. Autophagy
- •3.6. Necrosis
- •4. DEVELOPMENTAL CELL DEATH IN ZEBRAFISH EMBRYOS
- •5. THE P53 PATHWAY
- •6. PERSPECTIVES AND FUTURE DIRECTIONS
- •SUGGESTED READING
HOST–PATHOGEN INTERACTIONS |
377 |
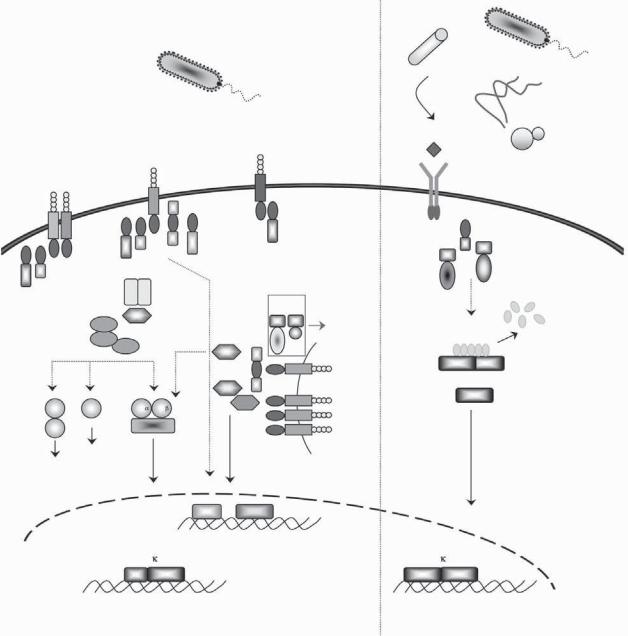
activated by Gram-negative bacteria and through Relish induces the production of a number of different AMPs, including Diptericin. Whereas the Toll pathway shares significant homology with the TLR and interleukin (IL)-1R pathways in mammals, the Imd pathway is related to the mammalian tumor necrosis factor receptor (TNFR) and NOD pathways (Figure 32-6). This striking conservation between the fly and mammalian pathways points to a common ancestry of these immune mechanisms.
3.2. PRRs and inflammation
3.2.1. TLRs
The innate immune system was extensively studied in Drosophila with the aim of elucidating how fruit flies that lack adaptive immunity responded to infectious pathogens. The discovery of Drosophila Toll was followed by the cloning of a human homolog and its characterization as a PRR able to stimulate the NF- κB pathway.24 So far, 12 members of the Toll family have been identified in mammals and are referred to as the Toll-like receptors (TLRs). All known TLRs are type I transmembrane proteins consisting of an extracellular domain with leucine-rich repeats (LRRs) responsible for ligand detection and a cytoplasmic Toll/IL1R homology (TIR) domain essential for initiating signaling25 (Figure 32-7). TLRs function as homoor heterodimers and recognize a broad range of microbial components.26 Mouse genetics studies and investigation of TLR knockout mouse phenotypes demonstrated the essential role of these receptors in pathogen recognition. Mice with a point mutation in Tlr4 are hyporesponsive to LPS.27 TLR3−/– mice fail to recognize viral doublestranded RNA (dsRNA) and are defective in stimulating inflammatory and type I interferon (IFN) responses.28 By associating with other TLRs, TLR2 responds to a variety of bacterial molecules that include peptidoglycan, lipoproteins, and lipopeptides. TLR5 recognizes bacterial flagellin,8 TLR7 responds to synthetic compounds with potent antiviral and antitumor activities such as resiquimod (R848), and TLR9 senses CpG motifs in bacterial and viral DNAs.29 TLRs share common signaling determinants with IL-1R family members; their TIR domains interact with TIR-containing adaptors, including MyD88, TIRAP/MAL, TRIF, and TRAM.30 With the exception of TLR3, all TLRs signal through MyD88. TIRAP/MAL, TRIF, and TRAM are more specialized. TRIF is engaged in response to viral PAMPs downstream of TLR3 and TLR4, MAL interacts with TLR2 and TLR4, and
TRAM binds TRIF in the TLR4 complex. The multistep signaling cascade induced by TLR activation results in the production of antimicrobial effectors by means of the NF-κB, mitogen-activated protein kinases (MAPKs), and interferon regulatory factor IRF 5/3/7 pathways31 (Figure 32-7).
3.2.2. NLRs
NLRs are evolutionarily related to disease resistance or “R” proteins in plants. These are cytosolic proteins that contain a nucleotide-binding site (NBS) and leucinerich repeats (LRRs) and are often involved in the recognition of PAMPs and pathogen-induced host danger signals. Upon activation, R proteins elicit a hypersensitive or guard response, which induces antimicrobial proteins, cell wall modification, and programmed cell death.32 Although the defense systems in animals and plants evolved under selective pressure imposed by distinct infectious pathogens, they stayed remarkably conserved across the two kingdoms. Another level of similarity between the two systems is the role that Hsp90 and SGT1 play in stabilizing the NBS-LRR/NLR proteins and maintaining their conformation in an auto-repressed but activation-competent state.33
The first described mammalian NLR, Nod1, was identified in a screen aimed at finding Apaf-1–related proteins.34 Similar to Apaf-1, Nod1 possesses an N- terminal CARD domain, a central nucleotide binding and oligomerization domain (Nod), and a C-terminal agonist-binding domain. The latter is a WD-40 repeat domain in Apaf-1 that binds cytochrome c, whereas it is an LRR domain in Nod1 that senses bacterial peptidoglycan derivatives.35 In response to agonist sensing, NLRs oligomerize and activate inflammatory effectors. To date, there are 22 NLRs, including 14 Nlrp proteins (Nlrp1–14), 5 Nods, Ipaf, Naip5, and CIITA. Comparison of the NLRs reveals both structural and functional differences. The N-terminal effector domain is variable among NLRs; it is a CARD in Nod1 and Ipaf, two CARDs in Nod2, three BIRs in Naip, and a pyrin domain (PYD) in Nlrp1-14 (Figure 32-8).
3.2.3. The Nod signalosome
Despite the presence of a CARD in Nod1 and Nod2, these proteins do not engage caspases directly. Instead, they interact with a CARD-containing kinase known as RIP2. This assembles a Nod signalosome that recruits TRAF proteins, cellular inhibitor of apoptosis proteins cIAP1 and cIAP2, TAK1, TAB1 and TAB2, needed to activate the

378 MAYA SALEH
Mammalian Insect
|
|
TNF |
TNF |
TNF |
|
|
|
|
|
|
TNFRTNFRTNFR |
|
|
|
|||
|
DD |
DD |
|
DD |
DD |
DD |
|
|
|
|
|
|
|
|
|
||
Complex I |
DD |
|
|
|
|
DD |
|
|
|
|
|
|
|
|
|
|
|
|
|
TRADD |
|
TRADD |
|
|
||
|
RIP1 |
|
|
|
|
RIP1 |
|
|
|
|
TRAF2 |
TRAF2 |
|
|
|
||
|
bbbUUU |
RING |
|
RING |
bb |
U |
||
U |
b |
|
|
|
|
|
Ubb |
|
|
|
|
|
|
UUU |
|
||
bU |
|
|
|
|
b |
|
|
|
|
BIR BIR |
BIRCARD |
RING |
|
|
|||
|
|
|
cIAP1/2 |
|
|
|
||
|
|
TAB2 |
|
|
|
|
|
|
|
|
|
|
|
TAK1 |
|
|
|
|
|
IKK |
IKK |
|
|
|
||
|
|
|
NEMO |
|
|
|
||
|
|
P |
|
P |
|
|
|
|
|
|
I |
B |
UbUbUb |
|
|
|
|
|
|
|
|
|
Ub |
|
|
|
|
|
p50 |
|
|
RelA |
|
|
|
DISC |
|
Complex II |
|
|||||
DED |
DED |
DD |
DD |
DD |
TRADD |
RIP1 |
TRAF2 |
RING |
|
DED |
|
|
|
|
|
|
|
p20 |
|
|
|
|
|
|
|
|
DED |
FADD |
DD |
DD |
TRADD |
RIP1 |
TRAF2 |
RING |
|
DEDDED |
DD |
|||||||
p10 |
|
|
|
|
|
|
|
|
p20 |
|
|
|
|
|
|
|
|
Pro-caspase-8/10 |
|
|
|
|
||||
p10 |
|
|
|
|
|
|
|
|
Caspase-3/6/7 |
|
|
|
|
|
|
||
p10p10 |
|
|
|
|
|
|
|
|
p20 |
|
p20 |
|
|
|
|
|
|
Substrate processing
Apoptosis
DAP-type
Peptidoglycan
derivatives
PGRP
-LC
IMD
DD
BIR BIR BIRCARD RING
DIAP2
TAB2
TAK1
IKK Ird5
Kenny
Rel
Gram-negative
bacteria
dFADD
|
DED |
DD |
DED |
DED |
|
p20
Dredd
p10
Relish
ANK
Rel
|
NF B |
|
|
NF B |
p50 |
RelA |
|
Rel |
Rel |
Induction of survival and |
DNA fragmentation |
Induction of Diptericin and other |
||
inflammatory genes |
Chromatin condensation |
antimicrobial genes |
||
Figure 32-6. Schematic representation of the mammalian TNF and insect Imd pathways. In mammals, TNF induces an inflammatory response mediated via the assembly of a signaling complex at the TNF receptor level, known as complex I. Through the recruitment of the adaptor TRADD (TNF receptor associated protein with a death domain [DD]), the kinase RIP-1 (receptor-interacting protein 1) and the E3 ubiquitin ligases, TRAF2 (TNF receptor-associated factor 2), cIAP1 and cIAP 2 (cellular inhibitor of apoptosis proteins), the signal is transduced to the kinases TAK1, TAB2, and IKKs, resulting in the activation of the NF-κB and MAPK inflammatory pathways. In conditions promoting cell death, TRADD, TRAF2, and RIP-1 dissociate from complex I and recruit the DISC (death-inducing signaling complex), which is composed of FADD and caspases-8/10, to form complex II, which is necessary to oligomerize and activate the caspases and initiate the extrinsic apoptosis program. In Drosophila melanogaster, the Imd (immune deficiency) pathway is engaged in response to infection with Gram-negative bacteria. Peptidoglycan derivatives from the bacterial cell wall activate PGRP-LC receptors on the plasma membrane of fat body cells, which transduce the signal to the NF-κB protein Relish, which in turn induces the expression of antimicrobial peptide genes such as Diptericin. The fly Imd signal transduction pathway shares similarities with the mammalian TNF pathway. Common e ectors include a RIP1-like adaptor with a death domain (Imd), an IAP protein (DIAP2), the kinases TAK1 and TAB2, IKKβ (ird5), IKKγ (Kenny), and NF-κB (Relish). In addition, dFADD and Dredd (the homolog of caspase-8) play an essential role in the activation of Relish, a homolog of mammalian NF-κB p105. One main di erence between the two systems is that Dredd cleaves the inhibitory ankyrin domain of Relish, freeing its Rel transcription domain to translocate to the nucleus and activate target genes. See Color Plate 38.
HOST–PATHOGEN INTERACTIONS |
379 |
Mammalian
Gram-positive and Gram-negative
bacteria
Insect
|
|
Gram-positive |
|
|
bacteria |
- |
Lysine-type |
|
PGRP |
S |
|
|
Peptidoglycan |
|
derivatives
|
|
|
|
|
LPS |
||
|
Bacterial |
|
|
|
|
||
|
lipoproteins |
|
|
|
|
||
|
|
TLR1/6 |
|
|
|
TLR4 |
|
|
|
TLR2 |
TIR |
TIR |
TIR |
TIR |
|
TIR |
TIR |
TIR |
TIR |
|
Mal |
|
TRIF |
|
|
|
|
MyD88 |
|
|
|
MyD88 |
|
|
IRAK1 |
IRAK4 |
|
|
|
|
Mal |
|
|
|
|
|
|
TRAF6
TAB2
TAK1
TAB1
MKK3 |
MKK7 |
IKK IKK |
MKK6 |
|
NEMO |
|
|
JNK
p38
Fungi
Flagellin |
|
Spätzle |
|
|
|
||
|
|
|
|
|
|
||
TLR5 |
|
|
|
|
|
|
Yeast |
|
|
|
Toll |
|
|
|
|
|
|
|
|
|
|
|
|
TIR |
TIR |
|
TIR |
|
|
|
|
TIR |
|
|
dMyD88 |
TIR |
|
|
|
|
|
|
DD |
|
|
||
MyD88 |
|
|
|
DD |
|
||
|
|
|
|
|
|||
|
|
DD |
|
|
Pelle |
||
TRAM |
|
|
|
|
|
||
|
|
Tube |
|
|
|
||
|
|
|
|
|
|
|
|
|
FADD |
|
|
|
|
|
|
|
DD |
DD |
Apoptosis |
Cactus |
|
||
|
|
DED |
|
||||
TRIF RIP1 |
|
|
|
|
|
||
|
|
|
ANK |
|
|
||
TRAF6 |
|
|
|
|
|
|
|
TIR |
TIR |
TLR3 |
Dif |
Rel |
Rel |
Dorsal |
|
|
|
|
|
|
|||
TRAF3 |
|
|
Nucleic |
|
Rel |
|
|
TBK1 |
TIR |
TLR7 |
acids |
|
|
|
|
|
|
|
|
||||
|
TIR |
TLR8 |
|
|
|
|
|
|
TIR |
TLR9 |
|
|
|
|
|
Endosome
IRF3 |
IRF7 |
Induction of the interferon |
|
|
response |
NF B |
NF B |
p50 RelA
Induction of survival and inflammatory genes
Rel Rel
Induction of Drosomycin and other antimicrobial genes
Figure 32-7. A schematic representation of the mammalian TLR and insect Toll pathways. One important difference between the two systems is that, unlike TLRs, insect Toll is not a pattern recognition receptor (PRR). In response to infections with Gram-positive bacteria, fungi, and yeast, soluble PRRs are activated in the hemolymph, resulting in the processing of Spatzle,¨ a cytokine-like molecule that engages Toll. Downstream of Toll or TLR activation, there is a certain degree of similarity in the signal transduction pathways. See Color Plate 39.
proinflammatory NF-κB and MAPK pathways. Recently, a protein termed CARD9 has been reported as a new addition to the Nod signalosome. CARD9 interacts with the Nod-Rip2 complex and selectively mediates activation of the MAPK pathway downstream of Nod stimulation (36 and references therein). Similar to RIP1’s
activation at the TNFR level, polyubiquitination of RIP2 by cIAP1 and cIAP2 is key to the transduction of the signal from Nod proteins to NF-κB and MAPK.37 The Nod signalosome is negatively regulated by caspase-12, which was shown to interact with RIP2 and inhibit Nod signaling by blocking RIP2 ubiquitination.38

380 |
MAYA SALEH |
NOD1 |
|
|
CARD |
NBD |
|
NOD2 |
|
CARD CARD |
NBD |
|
|
NOD3/9/27 |
|
|
X |
NBD |
|
(NLRC3/X1/5) |
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
NALP1 |
|
|
PYD |
NBD |
FIIND CARD |
(NLRP1) |
|
|
|||
|
|
|
|
|
|
NALP2-14 |
|
|
PYD |
NBD |
|
(NLRP2-14) |
|
|
|
||
|
|
|
|
|
|
IPAF |
|
|
CARD |
NBD |
|
(NLRC4) |
|
|
|
||
|
|
|
|
|
|
NAIP |
BIR |
BIR BIR |
CARD |
NBD |
|
(NLRB) |
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
CIITA |
|
|
AD |
NBD |
|
|
|
|
|
|
|
APAF1 |
|
|
CARD |
NBD |
|
Ced4 |
|
|
CARD |
NBD |
|
|
|
|
|
|
|
|
|
|
|
|
|
ASC |
|
PYD |
CARD |
|
|
CARDINAL |
|
FIIND |
CARD |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Caspase- |
|
|
|
p20 |
p10 |
3/6/7 |
|
|
|
|
|
Caspase-8/10 |
|
DED DED |
p20 |
p10 |
|
|
|
|
|
|
|
cIAP1/2 |
BIR |
BIR |
BIR |
CARD |
RING |
|
|
|
|
XIAP |
BIR |
BIR |
BIR |
|
RING |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
TRAF2 |
|
RING |
|
TZ |
TZ |
|
CC |
MATH |
|
TRAF6 |
|
RING |
|
TZ |
TZ |
|
CC |
MATH |
|
|
|
|
|
|
|
|
|
||
RIP1 |
|
|
|
Kinase domain |
|
DD |
|
||
RIP2 |
|
|
|
Kinase domain |
|
CARD |
|
||
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
||
IKK / |
|
|
|
Kinase domain |
LZ |
HLH |
Nemo BD |
||
IKK |
|
|
|
|
CC1 |
|
CC2 |
LZ |
ZF |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
FADD |
|
|
|
|
|
|
DED |
DD |
|
TRADD |
|
|
|
|
|
|
|
DD |
|
TNFR1 |
|
EC |
|
EC |
EC |
EC |
TM |
DD |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Caspase-1 |
|
|
|
CARD |
|
p20 |
p12 |
|
|
Caspase-9 |
|
|
|
CARD |
|
p20 |
p10 |
|
|
Ced3 |
|
|
|
CARD |
|
p20 |
p10 |
|
|
|
|
|
|
|
|
|
|
|
|
Figure 32-8. E ectors of inflammation. The domain structures are shown. AD, transcriptional activation domain; CC, coiled-coil; EC, extracellular domain; FIIND, domain with function to find; HLH, helix-loop– helix domain; LZ, leucine zipper domain; MATH, meprinand TRAF-homology domain; NBD, nucleotidebinding domain; PDZ, protein domain named after the initial letters of PSD-95, Dlg, and ZO-1; TM, transmembrane domain; TZ, TRAF-type zinc-finger domain; ZF, zinc finger domain. See Color Plate 40.
3.2.4. The inflammasome
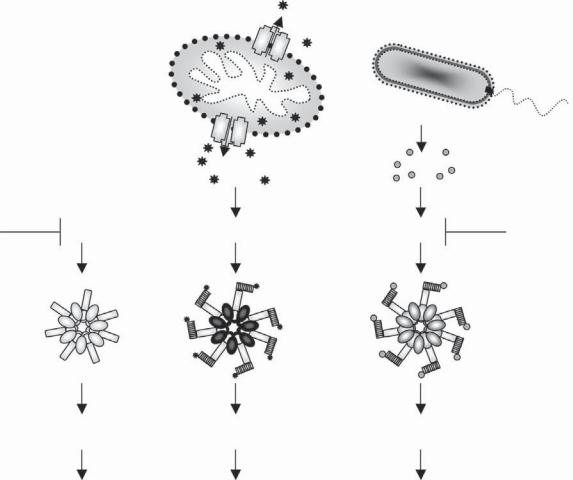
Unlike Nod proteins that signal through RIP2, NlRp proteins, Ipaf, and Naip recruit and activate inflammatory caspases, predominantly caspase-1, in a multiprotein complex known as the inflammasome.39,40 The activation of caspase-1 results in the processing and maturation of the cytokines IL-1β and IL-18. IL-1β and IL-18 exert various effects on different tissues, which result in the induction of fever, anorexia, fatigue, fat catabolism, secretion of acute-phase proteins, and activation of immune cells, leading to the release of other cytokines and chemokines.41 The recruitment of caspase-1 and its activation in the inflammasome mirrors that of caspase-9 in the apoptosome during mitochondrial apoptosis (Figure 32-9). Structurally, the NlRp1 inflammasome observed by cryoelectron microscopy also seems to share with the apoptosome its oligomeric nature.42 Binding of caspases to NLRs occurs through a CARD–CARD interaction, which is either direct, as in the case of Ipaf binding to caspase-143
and NlRp1 binding to caspase-5,44 or indirect, as in the case of PYD-containing NlRp proteins.44 Adaptor molecules mediate the association between caspases and NlRp proteins.40 Two adaptors have been identified, namely a PYD-CARD–containing adaptor termed Apoptosis-associated Speck-like protein containing a Caspase-recruitment domain (ASC) (also known as PYCARD), and Cardinal, a protein with homology to the C-terminal region of NlRp1. Among the inflammatory caspases, caspase-1 is universal to all inflammasomes and is the sole “effector” of cytokine processing. Caspase-5 is recruited to the NlRp1 inflammasome,44 where it acts as a caspase-1 cofactor, but does not appear to be involved in the NlRp3, Ipaf, and Naip5 complexes. Caspase-11, the murine ortholog of caspase-5, has also been suggested to act in vivo as an essential activator of caspase-1. In support of this, caspase-11–deficient mice were shown to be incapable of producing IL-1β and IL-18 in response to LPS stimulation.45 Nonetheless, it seems that the requirement for caspase-11 is not absolute but is restricted to certain stimuli, as caspase-1
HOST–PATHOGEN INTERACTIONS |
381 |
Cytochrome C |
PAMPs |
Ced9 |
Ced4 |
Apaf1 |
NLRs |
Bcl-2, |
|
|
|
|
Bcl-xl |
|
|
|
? |
|
|
? |
Apoptosome |
|
Inflammasome |
|
|
|
Active |
Active |
Active |
Ced3 |
Caspase-9 |
Caspase-1 |
APOPTOSIS |
APOPTOSIS |
PYROPTOSIS & |
|
|
INFLAMMATION |
Figure 32-9. A parallel between mitochondrial apoptosis and NLR innate immunity pathways. The apoptosome is sca olded by the protein Apaf-1 and assembles in response to cytochrome c release from the mitochondria during intrinsic apoptosis to activate caspase-9. NLR proteins that share a conserved structure with Apaf-1 sca old the inflammasome. During bacterial infection, bacterial products (PAMPs) or host-derived alarm signals arising during the infection stimulate the assembly of the inflammasome into an oligomeric complex that recruits and activates caspase-1. Activation of caspase-1 leads to pyroptosis and inflammation. A similar pathway exists in the nematode Caenorhabditis elegans, where Ced4, a homolog of Apaf1, oligomerizes with the caspase Ced3 to induce apoptosis. With the exception of the apoptosome, which is a heptameric oligomer, the number of proteins in the inflammasome and CED4 oligomers is hypothetical (question marks). It is also unknown whether PAMPs bind directly to NLRs. See Color Plate 41.
could be normally activated in the absence of caspase-11 in response to Listeria infection.46 Caspase-12 has been recently shown to act as an inhibitor of caspase-1.47 The extent of caspase-1 activation appears to vary among inflammasomes. Ipaf, as compared with NlRp3, activates caspase-1 more robustly. This results in rapid caspase- 1–dependent cell death, or pyroptosis (discussed below in 3.3.2), in addition to cytokine maturation and release. It has been recently proposed that, in response to K+ efflux, cells assemble one large ASC oligomer, or speck, per cell. Termed the pyroptosome, this platform is hypothesized to recruit most of the cellular caspase-1
off NLRs, leading to increased caspase-1 activation and cell death.48 In vivo findings, however, do not implicate ASC in pathogen-induced pyroptosis, as cell death proceeds normally in ASC-deficient cells, whereas it is inhibited in caspase-1−/– or Ipaf−/– cells. Both Ipaf and ASC are fully required for IL-1β production in response to Salmonella, Pseudomonas, or Legionella infection, yet only Ipaf-deficient macrophages are fully resistant to pyroptosis induced by these pathogens, whereas ASCdeficient cells are only partially protected.43,49,50,51
The presence of LRRs in NLR proteins is thought to mediate recognition of PAMPs by these cytosolic