

Глава 6 синдромы расстройств гемостаза [коагулопатии, двс-синдром]
Хотя интерес к процессам свертывания крови и гемостазу проявляется в медицинском мире издавна, учение о закономерностях коагуляции крови и ее патологии было разработано лишь в последние два-три десятилетия, когда были открыты факторы коагуляции и функции участников этого процесса — тромбоцитов, белков и др.
6.1. Физиология гемостаза
Гемостаз обеспечивает прекращение кровотечения из поврежденных сосудов. Процесс гемостаза сложен и состоит из нескольких последовательных стадий. Это прежде всего локальная вазоконстрикция в месте повреждения сосуда, развивающиеся затем специфические изменения функционального состояния тромбоцитов близ места повреждения, их агрегации, а также адгезии к эндотелиальной стенке сосуда в месте повреждения, к коллагеновым структурам и нитям в этой зоне и образование тромбоцитарного конгломерата (первичный гемостаз). На последующих этапах гемостаза (вторичный гемостаз) активируется каскадный процесс образования фибринового сгустка, укрепляющего и пропитывающего тромбоцитарный конгломерат, и фибринолитический процесс, направленный, с одной стороны, на ограничение самоподдерживающегося, цепного процесса гемокоагуляции, с другой — на лизис (растворение) гемостатической пробки, сыгравшей свою гемостатическую роль. После образования фибринового сгустка начинается активация фибробластических процессов.
Вазоконстрикция, развивающаяся немедленно после повреждения сосудистой стенки или циркулярного повреждения сосуда, является непродолжительным (обычно не более 1 мин) процессом и оказывает незначительное влияние на интенсивность кровоизлияния из сосуда. Под влиянием вазоконстрикции просвет сосуда уменьшается, но не более чем на 1/4—1/4 исходного диаметра.
Механизмы вазоконстрикции окончательно не выяснены. Вместе с тем известно, что главную роль в этом играет нейрогенное сокращение мышечных и клеточных элементов сосудистой стенки с участием гуморальных субстанций, выделяющихся из активированных тромбоцитов, таких как серотонин и тромбоксан A2.
Тромбоциты. В неповрежденном сосуде, т. е. в отсутствие структурных нарушений эндотелиальной поверхности сосудистой стенки, активации кровяных компонентов коагуляции крови не происходит.
После повреждения сосуда тромбоциты приклеиваются к обнаженным субэндотелиальным структурам [Добровольский В. И., и др., 1984]. Адгезия тромбоцитов усиливается также фактором Вилленбранда, представляющим собой высокомолекулярный плазменный белковый компонент молекулы фактора VIII. Точный механизм влияния фактора Виллебранда на адгезию тромбоцитов пока не ясен, однако известно, что при болезни Виллебранда (дефицит этого фактора) адгезия тромбоцитов замедляется или даже отсутствует, а время кровотечения увеличивается.
В зоне повреждения сосуда эритроциты высвобождают аденозин-5'-дифосфат (АДФ), который дополнительно активируег тромбоциты и ускоряет процесс адгезии и агрегации. Этот процесс носит название «реакция освобождения» [Born G. V. et al., 1976]. АДФ — главный стимулятор адгезии и агрегации. С увеличением числа эритроцитов и с повышением вязкости, крови способность тромбоцитов к адгезии существенно усиливается. Это обстоятельство лежит в основе возросшей тромбо-тической наклонности у больных с истинной красной полиците-мией [Bougthon В, J. et al., 1977].
После адгезии к субэндотелиальным структурам тромбоциты теряют дискоидную форму и выпускают длинные псевдоподии, которые прикасаются к поврежденной поверхности сосудов. Есть основание считать псевдоподии специфическими рецепторами тромбоцитарной мембраны к физиологическим, активаторам агрегации (АДФ, тромбин и коллаген). Названные субстанции начинают контрактильный процесс высвобождением Са2+ из мест его хранения и активацией тромбоцитарного актомиозина и тромбастенина. Изменения конфигурации и формы поверхности мембраны тромбоцитов является основным моментом активации ими коагуляционных процессов в крови; [Schick P. К. et al., 1976].
В зависимости от обстоятельств судьба агрегированных тромбоцитов может быть двоякой: они могут потерять способность поддерживать агрегированное состояние и распасться, но могут также превратиться в необратимые большие сгустки. Повышенный уровень цитоплазматического Са2+ активирует фосфолипазу аз, которая способствует освобождению жирных, кислот из клеточных фосфолипидов. Из них наиболее важной в процессах гемокоагуляции является арахидоновая, так как она служит основным компонентом при формировании тромбоксана А2 — одного из мощнейших стимуляторов тромбоцитарной агрегации и вазоконстрикции [Machin S. J., 1983]. Затем, тромбоцитарные лакуны и альфа-гранулы секретируют эти субстанции в зону повреждения сосуда и образования будущего сгустка вместе с АДФ. Последний проявляет себя как химический медиатор, способствующий агрегации. Тромбоциты выделяют также локальный вазоконстриктор, серотонин, катехол-амины и митогенный фактор, стимулирующий экстренную пролиферацию гладкой мускулатуры в зоне повреждения сосуда. При этом продуцируется ряд лизосомных ферментов [Ross R. et al., 1974]. В процессах агрегации тромбоцитов участвуют также другие субстанции белковой природы, например бета-тромбоглобулин, биологическая активность которого мало изучена, и фактор 4 тромбоцитов. Обе эти субстанции могут быть определены радиоиммунным методом [Ludlam С. А., 1975; Moore S. et al., 1975].
Другой адтиватор агрегации, способствующий усилению «реакции освобождения» АДФ и тромбоцитарных факторов,— это тромбин [Кузник Б. И. и др., 1971; Баркаган 3. С., 1980, и др.]. Более подробно о нем говорится дальше.
Плазменные факторы коагуляции. Изучение поражающего воображение процесса образования сгустка крови длится уже более 100 лет. Первоначально исследование этого вопроса было связано с именами русского ученого профессора Юрьевского университета А. А. Шмидта (1895), указавшего, что суть процесса образования сгустка состоит в превращении под влиянием фермента растворимого фибриногена в нерастворимый фибрин, и чешского ученого П. Моравитца (1905), который расшифровал и обобщил ход процесса образования фибрина и указал на роль в этом процессе образующегося в крови фермента тромбина. Была создана общая схема превращения фибриногена в фибрин под влиянием тромбина, образующегося в свою очередь в результате действия фермента тромбокиназы (известной теперь как фактор Ха) на протромбин.
В общих чертах она справедлива и в настоящее время.
В дальнейшем, главным образом в 50-х годах XX столетия, было открыто множество других участников процесса гемокоагуляции. В результате в 60—70-х годах удалось построить схему коагуляционного процесса, в которой нашли место все известные к этому времени факторы [McFarlan R., 1956]. Прежде чем рассмотреть современные схемы осуществления коагуляции крови, остановимся на номенклатуре участников этого процесса.
Номенклатура. Для обозначения плазменных факторов коагуляции в Международной номенклатуре (согласно решению Международной комиссии по тромбозу и гемостазу) используют римские цифры, которыми обозначают неактивированное состояние фактора, т. е. профермент (табл. 6.) Активированное состояние фактора (фермент) обозначают добавлением к римской цифре буквы «а», например: фактор II — протромбин, фактор Па—тромбин. Для обозначения тромбоцитарных факторов используют арабскую нумерацию, например: «фактор 4 тромбоцитов». Иногда факторы обозначают словесно, в основном по месту, где они образуются, или по их основной функции, например тканевый тромбопластин (фактор Ш),аксе-лерирующий фактор (V), фибринстабилизирующий фактор (XIII) и т. д. Встречаются также собственные имена (фамилии) в обозначении факторов, например: фактор Стюарта — Прауэра (X), фактор Хагемана (XII), фактор Кристмаса (IX). При этом используют фамилии больных, у которых впервые был обнаружен генетический дефицит обозначаемого фактора. Могут встретиться также упоминания о факторах Фитцжеральда (высокомолекулярный кининоген) и Флетчера (прекалликреин), которые не получили цифрового обозначения. Они являются компонентами общих ферментных систем, например калликреин-кининовой, но как аутосомные компоненты входят в антигемофильный глобулин (фактор VIII).
Основой коагуляционного механизма, состоящего из многокомпонентных ферментных и проферментных систем, кофакторов и ингибиторов, является последовательная ступенчатая активация процесса двумя основными путями: так называемым внутренним путем (механизмом), который начинается контактом факторов свертывания крови с поврежденной отрицательно заряженной поверхностью сосудистого эндотелия, и внешним путем (механизмом), начинающимся с освобождения тканевого тромбопластина из поврежденных клеток. Этот сложный ферментативный процесс ведет к образованию фибрина, оседающего затем на тромбоцитарной пробке и образующего тромб. Весь процесс образования фибринового сгустка отражен на схеме 6.1.
Таблица 6.1. Номенклатура плазменных факторов гемокоагуляции
Фактор |
Название фактора, синонимы |
I |
Фибриноген |
II |
Протромбин |
III |
Тканевый тромбопластин, тканевый фактор |
IV |
Кальций |
V |
Акселерирующий фактор, проакцелерин, Ас-глобулин, лабильный фактор |
VI |
Присвоенный номер и употребляется редко, фактор обладает акселерирующим эффектом, активатор V |
VII |
Проконвертин, стабильный фактор |
VIII |
Антигемофильный глобулин А |
IX |
Плазменный компонент тромбопластина (РТС-фактор), антигемофильный фактор В, фактор Кристмаса X ' Протромбиназа, фактор Стюарта — Прауэра |
XI |
Плазменный предшественник тромбопластина (РТА-фактор) |
XII |
Фактор Хагемана, контактный фактор |
XIII |
Фибриназа, фибринстабилизирующий фактор, плазменная трансглутаминаза |
Схема 6.1. Механизмы свертывания крови1
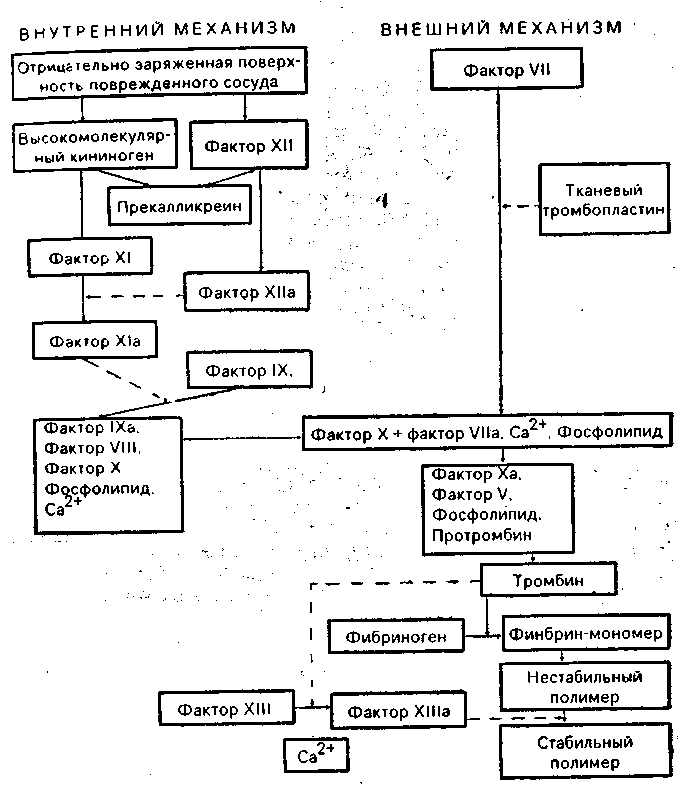
Сплошными стрелками обозначено превращение, пунктирными — активация
Внутренний путь процесса активации образования сгустка начинается с того, что комплекс, состоящий из фактора XII и высокомолекулярного кининогена, связанных с помощью прекалликреина [Веремеенко К. Н., 1977], адсорбируется на поверхности отрицательно заряженной поврежденной поверхности эндотелия, активирует фактор XI [Thompson R. Е., 1977]., превращая его из профермента в фермент (Х1а). Затем следует серия превращений инертных предшественников (фактор IX + фактор VIII, активированные фосфолипидом и Са2+) в активные сериновые протеазы, ведущие к формированию в конечном счете фибрина.
Появление в результате повреждения сосудистой стенки ряда тканевых субстратов (клеточные соки) активирует внешний путь коагуляции крови. Процесс начинается с конверсии фактора VII под влиянием тканевого тромбопластина в фактор Vila, т. е. в фермент, который в комплексе с Са2+ и фосфолипидами активирует затем фактор X. Участие фосфолипидов в процессах активации фактора X происходит с обеих сторон — как по внешнему, так и по внутреннему пути (см. схему 6.1). Происхождение фосфолипидов преимущественно тромбоцитарное: на поверхности тромбоцитов в зоне повреждения образуются мицеллы, содержащие фосфолипиды. Фосфолипиды ускоряют процесс активации фактора IX и превращения его из профермента в фермент (1Ха). Последний в комплексе с анти-гемофильным глобулином (VIII), Са2+ и фосфолипидами активирует фактор X.
Как внешний, так и внутренний механизмы и пути каскадно-комплексных превращений направлены на активацию одного из центральных компонентов коагуляционного процесса — фактора X, который затем в комплексе с акселелирующим фактором V, Са2+ и тромбоцитарными фосфолипидами превращает протромбин в тромбин (см. схему 6.1). В норме в плазме крови тромбина нет. Он появляется лишь тогда, когда в результате повреждения сосудистой стенки запущен механизм образования сгустка крови.
Наблюдения за больными, страдающими наследственным дефицитом или недостаточностью фактора X (болезнь Стюарта—Прауэра), показали, что отсутствие в крови этой сериновой протеазы блокирует оба (внешний и внутренний) пути активации механизма свертывания крови, в результате чего становится невозможным образование протромбиназной активности. Однако в норме активированный фактор X, сам по себе обладает довольно слабой протромбиназной активностью. Его эффект повышается буквально в тысячи раз акселелирующим фактором V (возможно, при содействии акселерирующего фактора VI). Аналогично этому активирующее влияние фактора IX на фактор X на 3—4 порядка усиливается действием антиге-мофильного глобулина (фактор VIII). На этом основании в последнее время факторы свертывания крови принято делить на две большие группы — ферментную, к которой относятся факторы XII, XI, IX, VII, X и II, и неферментную — факторы I, V и VIII [Баркаган 3. С., 1980].
Схема 6.2. Процесс фибринолиза
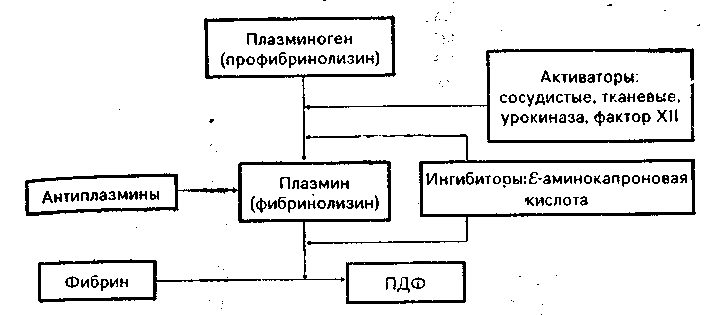
Под влиянием протромбиназы (тромбокиназы, фактора Ха) циркулирующий в крови протромбин превращается в тромбин, который в свою очередь является фактором превращения фибриногена в фибрин. Процесс осуществляется путем последовательного расщепления циркулирующего или местного фибриногена на отдельные фрагменты — пептиды. От каждой а- и бета-цепи фибриногена отщепляются по два фибринопептида (пептиды А и пептиды В). Оставшиеся фибрин-мономеры с четырьмя свободными связями соединяются между собой в полимер, из которого формируются нити — волокна фибрина. Однако фибрин-полимер — неустойчивое соединение и под влиянием различных факторов может быть вновь расщеплено на мономерные фрагменты. Укрепление полимеризованного фибрина происходит под влиянием фибринстабилизирующего фермента трансглутаминазы (фактор XIII), которая «прошивает» фибрин-полимер дополнительно по гамма-цепям и делает его стабильным, устойчивым к действию белковых растворителей, мочевины и других сред [Балуда В. И., 1977].
Фибринолитическая система. Растворение фибрина зависит от активации фибринолитической ферментной системы [.Davidson J. F., 1977]. Плазминоген циркулирует в крови как неактивный профермент (в норме концентрация 110—240 мг/л) и под действием активирующих факторов превращается в активную сериновую протеазу плазмин. По характеру протеолитического действия плазмин близок к трипсину. Процесс активации фибринолитической системы представлен на схеме 6.2, из которой следует, что возможно несколько неодинаковых по значению путей активации плазминогена и перевода его в плазмин.
Главным физиологическим активатором плазминогена, освобождающимся из клеток сосудистого эндотелия и действующим постоянно, является сам сосудистый эндотелий. Стимулами к высвобождению сосудистого активатора плазминогена являются вазоактивные соединения, окклюзия венозных сосудов, повышение температуры тела, физические упражнения и др. Тканевый активатор плазминогена в больших количествах содержится в матке, предстательной железе, легких и в нормальных условиях в кровь не попадает. Но в условиях травмы и при образовании фибрина его локальная концентрация может резко возрастать. Клетки почечной паренхимы образуют и высвобождают в кровоток специфический активатор урокиназу, которая в нормальных условиях экскретируется в мочу. Наконец, мощнейшим активатором плазминогена является фактор XII, а точнее, весь комплекс инициации внутреннего механизма коагуляции крови, включая фактор XII, кинин-калликреиновую систему и систему комплемента [Stormorken H., 1977].
На примере роли фактора XII в инициации фибринолиза можно показать, что запуск основных механизмов коагуляции крови сам по себе предполагает и запуск фибринолитйческого механизма, т. е. можно установить единство коагуляционной и фибринолитической систем.
В норме физиологическая концентрация плазменных активаторов довольно быстро снижается при помощи естественных антиактиваторов, а также путем метаболизма (деградации) в печени. Только в месте образования тромба плазминоген превращается в плазмин в необходимых количествах. Часть плазмина, поступившего в общую циркуляцию, быстро инактивиру-ется с помощью антиплазминов [Collen D., Wiman В., 1978]. Плазмин превращает фибрин в продукты его расщепления и таким образом растворяет сгусток; в этом его единственное назначение.
Ингибиция тромботической активности. Для предупреждения неконтролируемой активности большого числа взаимодействующих реакций, ведущих к образованию фибринового сгустка, в организме существует мощная система естественных ингибиторов коагуляции. Главный из них антитромбин-III, основное назначение которого — тормозить образование и активность тромбина. Он ингибирует также эффект активированных факторов IХа, Ха, ХIа, калликреина и плазмина, [Seegers W. Н., 1978]. В нормальных условиях гепарин связывается с антитромбином-III, изменяет его молекулярную конфигурацию и таким образом усиливает и ускоряет ингибирующую активность антитромбина-III в отношении тромбина и фактора Ха [Rosenberg R. D., 1978]. Важными ингибиторами тромбина являются О2-макроглобулин и а2-антитрипсин. Оба эти ингибитора способны воздействовать как на внешнем, так и на внутреннем пути коагуляционного процесса.
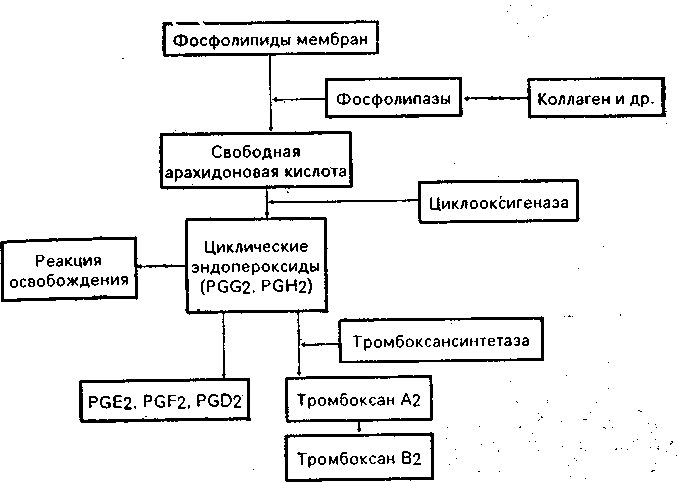
Схема 6.3. Метаболизм тромбоцитарной арахидоноеой кислоты
В 1976 г. J. Stenflo обнаружил в плазме крови крупного рогатого скота новый витамин-К-зависимый белок, который был назван протеином С. Механизм его действия достаточно изучен в последние годы. Хотя по структуре и функциям он признан аналогичным аутопротромбину АИ, его выраженный антитромботический эффект позволяет поставить его в ряд таких мощных антикоагулирующих факторов, как антитромбины. Специфический антикоагулянтный эффект протеина С проявляется удлинением протромбинового и активированного частичного тромбопластинового времени и обусловлен в основном инактивацией факторов V и VIII, главным образом их активированных форм. У больных в критических состояниях, а также у оперированных больных концентрация протеина С в плазме крови снижается обычно на 20% [Рябов Г. А. и др., 1989]. Врожденный дефицит протеина С сопряжен обычно с развитием тромботических осложнений.
Длительное присутствие тромбина в кровотоке ведет к потреблению протеина С и снижению его содержания в плазме. При развернутой картине ДВС-синдрома содержание протеина С снижается до 20—70% нормы.
Следует сказать несколько слов об ингибиции тромбоцитарной активности. Нормальные эндотелиальные клетки способны синтезировать из арахидоновой кислоты и высвобождать в кровоток простациклин (схема 6.3; страница 224), который является мощным вазодилататором и наиболее мощным естественным тромбоцитарным антиагрегантом [Varie J. R., Bergstrom S., 1979], Простациклин, который также продуцируется в легких, связан со специфическими рецепторами мембраны тромбоцитов и активирует связанную с мембраной аденилатциклазу, которая в экстремальных состояниях продуцирует повышенное количество цАМФ. Последний тормозит агрегацию тромбоцитов, замедляет реакцию освобождения и синтез тромбоксана Аг. Таким образом, сама сосудистая стенка контролирует избыточную активность тромбоцитов путем регуляции продукции простациклина, а также локально активирует фибринолитическую систему путем высвобождения активаторов плазминогена из эндотелиальных клеток. Эта реакция выражена преимущественно в тех случаях, когда образуется избыточное количество фибрина. Следует особо подчеркнуть взаимодействие сосудистого и плазменного механизмов в интеграции всего коагуляционного процесса.
Клинические и лабораторные проявления гемостатических расстройств. Взаимоотношения между образованием сгустка и лизисом в системе гемокоагуляции строго сбалансированы. В нормальном здоровом организме практически невозможен процесс бесконечного образования свертка, так же как невозможен непрерывный его лизис. Однако при заболеваниях, в частности при критических состояниях, наблюдается множество отклонений от нормального баланса в ходе гемостаза, которые могут проявляться либо кровотечением, либо, наоборот, повышенным тромбообразованием. Следовательно, необходима точная диагностика этих отклонений.
Первой проблемой при лечении больного с кровотечением является необходимость определить, является ли оно локальным, например при язве желудка, или представляет собой результат общих расстройств гемокоагуляции. Мы рассмотрим только нарушения гемокоагуляции и не будем касаться проблем диагностики хирургических, гинекологических или акушерских кровотечений. Принципиально патогенез гемостатической недостаточности и кровотечение могут быть обусловлены следующими основными причинами: тромбоцитопенией, функциональной патологией тромбоцитов, патологическим состоянием сосудистой стенки, дефицитом или дефектом факторов коагуляции, повышенным фибринолизом и сочетанием нескольких или всех указанных причин.
При оценке характера кровотечения следует учитывать не только данные лабораторных исследований; но также анамнез и клиническую картину.
Длительное кровотечение после экстракции зубов или операций может указывать на дефекты гемостаза, но не является специфичным для какого-либо типа его расстройств: оно может быть связано как с дефицитом и расстройством функциональной активности тромбоцитов, так и с дефицитом факторов свертывания крови или повышением фибринолитической активности.
Экхимозы и петехии могут свидетельствовать о нарушении функции тромбоцитов или о наличии острого сосудистого стаза. Носовые кровотечения чаще бывают при нарушении функции капилляров (в: частности, при авитаминозах), но могут встречаться и при острых тромбоцитарных расстройствах. Пурпура и меноррагия наиболее часто возникают при болезни Вилле-бранда, тромбоцитопении и дисфункциях тромбоцитов, а также при повышенной чувствительности к производным ацетилсалициловой кислоты. Спонтанные гемартрозы и глубокие мышечные гематомы практически всегда указывают на возможность гемофилии. Развитие у боль-ного множественных кровотечений из мест уколов, операционных разрезов, трахеостомы, при сопутствующем кровотечении из поврежденных слизистых оболочек свидетельствует о развитии ДВС-синдрома. Однако этот синдром — состояние практически всегда ситуационное, т. е. обусловленное каким-либо предшествующим патологическим явлением, например кровотечением, сепсисом, распространенным опухолевым процессом и др.
Для установления точной причины клинически определяемых расстройств гемостаза необходима постановка специальных лабораторных тестов. Вначале целесообразно использовать простейшие скрининговые тесты, с помощью которых можно облегчить диагностику. Наиболее важные из них могут быть сгруппированы следующим образом:
1) для оценки тромбоцитарных расстройств: число тромбоцитов, время кровотечения, исследование мазка;
2) для оценки капиллярных расстройств: турникетная проба, время кровотечения;
3) для выявления расстройств гемокоагуляции: протромби-новое время; активированное частичное тромбопластиновое время; тромбиновое время, концентрация фибриногена.
Каждая лаборатория имеет собственные нормы по каждому из названных тестов. Если скрининговые тесты указывают на отклонения от нормы, то необходимо провести более подробное исследование показателей тестов, факторов коагуляции крови, качества тромбоцитов, состояния фибринолитической и антитромбиновой систем. Рассмотрим их подробнее.