

Актуальные вопросы современной гепатологии
.pdfРис. 2. Морфологические ультраструктурные изменения механизма некроза в процессе гибели клетки (Rosser B., Gores G., 1995).
Кроме влияния продуктов аутолитического распада печени и патологических метаболитов, накапливающихся у больных с ПЭ, важную роль в развитии некротического процесса в печени играют расстройства органного кровообращения и микроциркуляции, а также тесно связанная с ними циркуляторная гипоксия.
Основные стадии некробиотических изменений хорошо изучены (см. рисунок). Клетка претерпевает определенные изменения: появляется цитоплазматическая складчатость, набухают органеллы, пикнотизируется хроматин, клеточные органеллы концентрируются вокруг ядра и, наконец, происходят лизис цитоплазматической оболочки и гибель клеточных органелл (митохондрии, лизосомы и т.д.). Как правило, вокруг гибнущих клеток возникает воспаление, которое затем трансформируется в фиброзные изменения.
Ключевые патогенетические механизмы печеночной энцефалопатии
В развитии ПЭ могут принимать участие два основных механизма:
1)Наличие тяжелого острого или хронического заболевания печени, сопровождающегося выраженным снижением ее детоксицирующей функции (обусловливает развитие «эндогенной» или «истинной» ПЭ).
2)Формирование функциональных или анатомических шунтов обусловливает развитие портосистемной энцефалопатии (ПСЭ).
Эндогенная ПЭ в «чистом» варианте развивается при фульминантной печеночной недостаточности (ФПН) как результат гибели большей части паренхимы печени. В основе ПЭ при циррозе лежит
141
ПСЭ, хотя определенная роль принадлежит и снижению способности печени к обезвреживанию кишечных токсинов. Знание патогенетических факторов ПЭ важно для выбора оптимального метода лечения. Так, к патогенетическим факторам печеночной энцефалопатии относят:
1.Эндогенные нейротоксины
2.Аминокислотный дисбаланс
3.Нарушение баланса нейротрансмиттеров
4.Изменения постсинаптических рецепторов
5.Нарушение функционирования гематоэнцефалического барьера.
Многолетнее изучение патогенеза ПЭ позволяет прийти к выводу, что, по всей видимости, рассмотренные механизмы действуют в комплексе. Комбинация неблагоприятных факторов у больного с острой или хронической патологией печени ведет к развитию комплекса нервно-психических нарушений, обозначаемых как печеночная энцефалопатия.
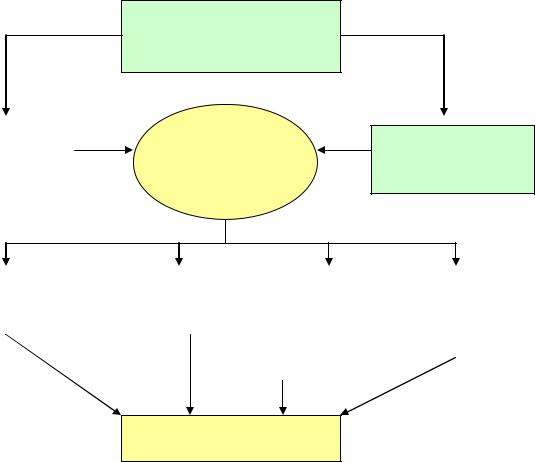
Нарушение функции печени или портосистемное шунтирование крови
Аминокислотный |
Отек и |
|
|
дисбаланс |
функциональные |
|
изменения |
|
|
|
астроглии |
Эндогенные
нейротоксины
Изменение |
|
Изменение |
|
Изменение |
|
Снижение |
постсинаптических |
|
Процесса |
|
проницаемости |
|
обеспечения |
рецепторов |
|
нейротрансмиссии |
|
гематоэнце- |
|
нейронов |
|
|
|
|
фалического |
|
энергией |
|
|
|
||||
|
|
|
|
барьера |
|
|
|
|
|
|
|
|
|
Клинические проявления печеночной энцефалопатии
Рис. 3. Схема патогенеза печеночной энцефалопатии
(Haussinger D., Maier K. Hepatische Enzephalopathie.- Verlag, 1996).
142
Патогенез печеночной энцефалопатии, несмотря на интенсивные исследования в этой области, до конца не изучен. Его уточнение существенно замедляют следующие обстоятельства:
-должны рассматриваться два различных причинновзаимосвязанных уровня – печень и головной мозг;
-посмертные исследования мозга при ПЭ проводятся редко и малопригодны для выяснения причин функциональных изменений;
-результаты экспериментов на животных с трудом можно перенести на человека;
-ПЭ является функциональным, динамичным и обратимым состоянием, каждый результат которого характеризует лишь небольшой промежуток времени.
Теория «отека астроглии».
Среди исследователей, изучающих эту проблему, нет единого мнения о приоритетности факторов патогенеза ПЭ. Многие из них придерживаются теории «отека астроглии», согласно которой печеночноклеточная недостаточность и/или портокавальное шунтирование, сопровождающиеся аминокислотным дисбалансом, приводят
кувеличению содержания нейротоксинов в ЦНС. В результате чего возникают отек и функциональные нарушения астроглии, которая играет принципиальную роль в трофике нейронов и процессах нейротрансмиссии.
Использование в последние годы в клинической практике более информативных методов исследования головного мозга позволило верифицировать морфологический субстрат повреждения ЦНС при печеночной энцефалопатии. Таким образом, у больных с ПЭ инструментально зафиксирован феномен отека астроглии, приводящий
ксерьезным функциональным последствиям. Несмотря на отсутствие клинических признаков повышения внутричерепного давления, четко регистрируются нарушения системы, поддерживающей осмотическое равновесие системы астроцит-нейрон, повышение проницаемости гематоэнцефалического барьера, изменение активности ионных каналов, снижение обеспечения нейронов АТФ и нарушение процессов нейротрансмиссии.
Изменение содержания осмотически активных веществ в астроцитах приводит к формированию гипергидратации астроцитов, то есть отеку мозга – центральному звену патогенеза ПЭ. Развитию гипергидратации астроцитов также способствуют бензодиазепины,
143
ГАМК-содержащие препараты, диуретики, некоторые цитокины (фактор некроза опухоли альфа). Аналогично патогенетическое действие таких разрешающих факторов, как кровотечение, инфекции, электролитный дисбаланс. Степень гидратации астроцитов обуславливает клиническую тяжесть и возможную обратимость церебральных нарушений.
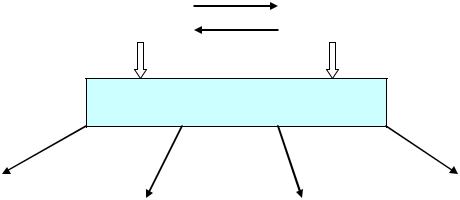
Аминокислотный |
|
Эндогенные |
дисбаланс |
|
нейротоксины |
|
|
|
ОТЕК АСТРОГЛИИ
Повышение |
|
|
|
|
|
Снижение |
проницаемости |
|
Изменение ак- |
|
Нарушение про- |
|
обеспечения |
гематоэнцефа- |
|
|
|
нейронов |
||
|
тивности ионных |
|
цессов нейро- |
|
||
лического барь- |
|
|
|
АТФ |
||
|
каналов |
|
трансмиссии |
|
||
ера |
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Рис.4. Патогенез печеночной энцефалопатии.
Наиболее известны также еще три теории патогенеза ПЭ: теория токсического действия аммиака и эндогенных нейротоксинов; теория ложных нейротрансмиттеров и теория нарушения обмена γ
– аминомасляной кислоты (ГАМК). Первая из них наиболее старая в историческом плане (С.С. Салазкин, 1898 год) и в то же время - наиболее обоснованная.
Теория токсического действия аммиака.
В древней Греции и в средние века врачи уже имели некоторое представление о печеночной недостаточности. Однако только после работ, проведенных Conway и Seligson в 1940 году по количественной оценке аммиака в крови, стало понятно, что состояние энцефалопатии, которое называлось физическим отравлением организма, развивается вследствие высоких концентраций аммиака в крови. Другим интересным аспектом в изучении этого патологического состояния является предоставление подробных отчетов патологоанатомических исследований пациентов, умерших с наличием симптомов энцефалопатии и высоким содержанием аммиака. Эти
144
отчеты были сделаны Иносэ, а данное состояние было названо гепатогенной энцефалопатией Иносэ. Несмотря на все эти успехи, патологическим изменениям в печени в то время не придавалось особого значения.
В настоящее время считается, что среди эндогенных нейротоксинов ведущее место отводится аммиаку (NH3). В организме постоянно поддерживается равновесие между его образованием и обезвреживанием. Важнейшими органами образования аммиака являются: толстая кишка – в результате гидролиза белка и мочевины интестинальной уреазоположительной микрофлорой; мускулатура
– пропорционально физической нагрузке; почки – продукция аммиака в которых существенно повышается при гипокалиемии и алкалозе; тонкая кишка – при распаде глутамина, основного источника энергии клеток слизистой оболочки (рис. 5).
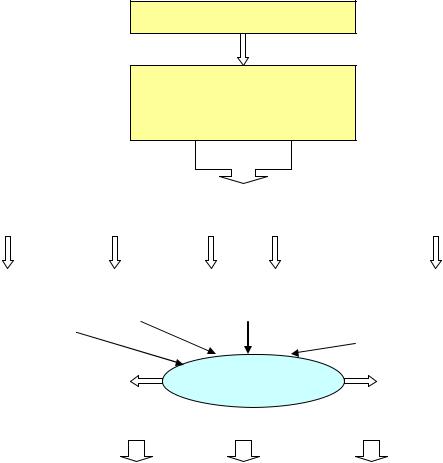
Образование аммиака
Белок и другие азотсодержащие субстанции пищи
Печень |
|
|
Почки |
|
|
Мышцы |
|
|
Тонкая |
|
|
|
Толстая |
|
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
кишка |
|
|
|
кишка |
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
Дезаминиро- |
|
|
Гипокалиемия |
|
|
|
Распад |
|
|
|
|
|
Гидролиз белка и |
|
|||||||||
|
вание |
|
|
|
алкалоз |
|
|
|
глутамина |
|
|
|
|
|
мочевины уреазо- |
|
|||||||
аминокислот |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
положительными |
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
бактериями |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
Аммиак |
|
|
|
|
|
||||||||||
|
Выделение в газо- |
|
|
|
|
|
|
|
|
|
Выделение в виде |
|
|||||||||||
образном состоянии |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
NH4 + с мочой и |
|
||||||
|
через легкие |
|
|
Обезвреживание аммиака |
|
|
|
фекалиями |
|
||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
Обезвреживание |
|
|
|
|
|
Образование |
|
||||||
|
Орнитиновый |
|
|
|
|
|
|
|
|
|
|||||||||||||
|
|
|
|
|
и элиминация |
|
|
глутамина: печень, |
|
||||||||||||||
|
цикл: печень |
|
|
|
|
|
|
|
|||||||||||||||
|
|
|
|
|
в кишечнике |
|
|
мышцы, астроциты |
|
||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Рис. 5. Обмен азота и аммиака
145
В тканях и жидкостях аммиак существует в виде иона аммония NH4 + в равновесии с небольшой концентрацией неионизированного аммиака NH3. Содержание аммиака в организме человека невелико, его концентрация в крови составляет 11-55 мкмоль/л. Нарушение баланса аммиака приводит к повышению его концентрации в крови и проявлению токсических свойств, не компенсируемых организмом.
Аммиак может выделяться только в небольшом количестве в виде иона аммония с мочой и фекалиями, а также в газообразном состоянии с выдыхаемым воздухом через легкие. Поэтому низкая концентрация аммиака в организме поддерживается с помощью механизмов, обеспечивающих его связывание.
Более 50% аммиака возникает при расщеплении белков в печени. Аммиак, образующийся в толстой кишке из продуктов распада белков под действием аммониегенной микрофлоры, поступает по воротной вене в печень, где большая его часть в норме включается в орнитиновый цикл, конечный продукт которого - мочевина. Этот цикл является важнейшим процессом связывания аммиака, происходящим в перипортальных гепатоцитах.
Рис.6. Биохимический цикл связывания аммиака и синтеза мочевины в печени.
Не включившийся в цикл мочевины NH3 захватывается небольшой популяцией перивенозных гепатоцитов, в которых из раз-
146
личных амино- и кетокислот (глютамат, α-кетоглютарат) и аммиака под влиянием глютаминсинтетазы образуется глютамин. Оба эти пути служат для предотвращения попадания токсичного аммиака в системный кровоток.
При печеночной энцефалопатии скорость метаболизма аммиака и других токсинов в печени значительно снижается. Кроме того, NH3 попадает в общий кровоток по портокавальным анастомозам и выключается из печеночного метаболизма.
Повышение уровня аммиака в крови приводит к его проникновению через гематоэнцефалический барьер в головной мозг, где он вызывает нейротоксический эффект: нарушается экспрессия генов, кодирующих ключевые белки астроцитов, снижается активность функции каналов хлора на мембране нейронов.
Механизм токсического действия аммиака на центральную нервную систему (ЦНС) до конца не ясен. Наиболее обоснованной является теория нарушения энергетических процессов в нейронах.
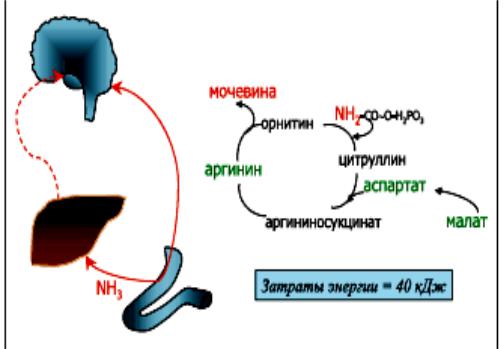
Неионизированный аммиак (его количество составляет 1 – 3 % от всего аммиака крови) легко проникает через гематоэнцефалический барьер, клеточные мембраны нейронов и мембраны митохондрий. В митохондриях идет ферментативная реакция, в результате которой из иона аммония и α-кетоглютарата образуется глютамин. Отмечается снижение скорости окисления глюкозы и образования АТФ в ткани мозга, что приводит к энергетическому голоданию клеток мозга, а усиленный синтез глютамина в мозге вызывает осмотический отек. Таким образом, происходит нарушение функции малатаспартатного челнока (рис.7).
При развитии печеночной энцефалопатии повышение концентрации аммиака колеблется обычно между 70 и 400 % по сравнению с верхней границей нормы. Повышение аммиака сыворотки крови регистрируется у 82 – 87 % больных с ПЭ.
(А.Г. Рахманова, 1983; С.Д. Подымова, 1997; С.Н. Соринсон,
1997.)
В норме степень аммониемии по методу Конвея равняется 28,6-85,8 мкмоль/л, а по технически несложному методу Мюллера
– Байзенхирца в модификации Энгельгардта 64,0 ± 14,3 мкмоль/л. Подтверждение участия аммиака в патогенезе ПЭ основано на
многочисленных данных:
- аммиакпродуцирующие вещества (белки, аминокислоты) могут вызывать ПЭ у больных циррозом печени, ограничение этих веществ (белково-редуцированная диета) улучшает или полностью устраняет ПЭ;
147
-врожденные дефекты синтеза мочевины, терапия глютаминазой или аспарагиназой, синдром Рейи сопровождаются гипераммониемией и дают нейропсихические симптомы, сходные с таковыми при ПЭ;
-прямая зависимость между степенью ПЭ и концентрацией аммиака в крови;
-степень ПЭ и концентрация глютамина в ликворе, отражающая содержание аммиака в мозге, коррелируют между собой.
АСТРОЦИТ
ЦИТОПЛАЗМА |
МИТОХОНДРИИ |
|
|
Оксалоацетат |
|
Малат |
|
Малат |
|
Оксалоацетат |
|
|
|
|
|
|
|
|
|
|
С1 |
|
||
|
|
|
|
|
||
|
a-кетоглутарат |
|
а-кетоглутарат |
|
||
|
|
|
|
|
|
|
|
|
Аспартат |
|
Аспартат |
|
|
|
|
|
|
|
|
С2 |
|
|
|
|
|
|
АММИАК |
|
Глутамат |
|
Глутамат |
|
|
|
|
|
ГЛУТАМИН
Рис.7. Нейротоксичность аммиака.
К группе эндогенных нейротоксинов относятся также меркаптаны, фенолы, индол, индикан, низкомолекулярные жирные кислоты. Аммиак может усиливать нейротоксический эффект меркаптанов и короткоцепочечных жирных кислот, концентрация которых повышена при болезнях печени.
148
Меркаптаны являются продуктами бактериального гидролиза серосодержащих аминокислот (метионин, цистеин, цистин) в толстом кишечнике. В норме они обезвреживаются печенью. При печеночной энцефалопатии концентрация меркаптанов в крови повышается, и они являются причиной печеночного запаха (foeter hepaticus). Хорошо известно разрешающее действие метионина на развитие ПЭ, а также уменьшение его токсичности посредством подавления антибиотиками интестинальной микрофлоры. Механизм действия меркаптанов связывают с увеличением транспорта ароматических аминокислот (триптофана, фенилаланина, тирозина) в головной мозг.
Жирные кислоты – это следующая группа нейротоксинов, образующихся из пищевых жиров под воздействием кишечных бактерий. У больных с ПЭ найдено повышение концентрации в крови жирных кислот с короткой цепью. К ним относятся: масляная, валериановая, капроновая, каприловая. Эти кислоты тормозят синтез мочевины в печени, чем способствуют гипераммониемии; вытесняют триптофан из связи с альбуминами, что приводит к увеличению потока триптофана в головной мозг. Все жирные кислоты воздействуют также на синапсы нервных клеток, замедляя проведение нервных импульсов. Особенно токсичны для головного мозга масляная и валериановая кислоты.
Нормальное содержание суммарного количества жирных кислот в сыворотке крови по методу В. Holmguist составляет 8,7 ± 3,5 мг%;
Фенолами обозначается группа дериватов тирозина и фенилаланина, также образующаяся в кишечнике под воздействием бактерий. Высокие концентрации фенолов приводят к развитию печеночной комы, четко коррелируя с глубиной ПЭ.
Нормальное содержание общих фенолов сыворотки крови (по
D. Mütting) 0,23 мг%.
Печень служит высокоэффективным "фильтром", проходя через который портальная кровь очищается от кишечных аутотоксинов. Благодаря химическим превращениям, реализующимся в печеночных клетках (гидроксилирование, дезаминирование, деметилирование, ацетилирование, образование парных соединений) все эти эндогенные нейротоксины трансформируются в нетоксичные продукты, которые выводятся из организма.
149
Это один из важнейших механизмов защиты головного мозга. При выпадении нейтрализующей функции печени токсичные вещества не обезвреживаются, поступают в системный кровоток и вызывают интоксикацию. Поскольку к любым токсинам особенно чувствительна нервная ткань, нейроциты головного мозга, прежде всего выявляется церебротоксический эффект.
Теория ложных нейротрансмиттеров.
Наряду с эндогенными нейротоксинами другой важной причиной функциональных нарушений является аминокислотный дисбаланс, приводящий к синтезу ложных нейротрансмиттеров. Аминокислотный дисбаланс при ПЭ характеризуется увеличением уровней ароматических аминокислот: фенилаланина, тирозина, триптофа-
на и снижением содержания аминокислот с разветвленной боковой цепью – валина, лейцина, изолейцина. Коэффициент Фишера (валин + лейцин + изолейцин фенилаланин + тирозин + триптофан) в норме равный 3,0 – 4,5 , снижается при ПЭ до 1 и ниже.
Ферментные системы мозга оказываются не в состоянии обеспечить обычный метаболический путь повышенному потоку ароматических аминокислот.
Повышенное поступление в мозг ароматических аминокислот, являющихся предшественниками ложных нейротрансмиттеров, вызывает существенное торможение ферментов, превращающих тирозин в диоксифенилаланин (ДОФА). ДОФА должен преобразовываться в дофамин и норадреналин, но метаболизм этих исходных соединений протекает по альтернативному пути с накоплением в ЦНС ложных нейротрансмиттеров: октопамина, тирамина, фенилэти-
ламина. Они вытесняют из синапсов первичных окончаний истинные медиаторы – норадреналин и дофамин. Таким путем происходит дезорганизация работы нервной системы.
Участие аминокислотного дисбаланса в патогенезе ПЭ подтверждается повышенными концентрациями октопамина в крови и моче у больных ПЭ. Ложные нейротрансмиттеры и продукт метаболизма триптофана – серотонин, дающий ингибиторный эффект, вызывают угнетение нервной системы, истощение мозга и развитие энцефалопатии.
Большое значение в регуляции сна и поведении у человека имеет содержание серотонина и его рецепторов в центральной нервной системе. При тяжелых поражениях печени увеличивается концентрация триптофана в крови и головном мозге, что приводит
150