

4.4.3 Современная трактовка основных понятий координационной теории
За прошедшие годы настолько расширился круг известных (чаще синтезированных искусственно) веществ, настолько изменилось наше понимание об их строении, что стало довольно трудно провести четкую разграничительную линию между соединениями "комплексными" и более "традиционными". Нередко оказывается, что вещества, чьи формулы записываются очень просто и лаконично (Al2O3, ZnS, CoCl2), устроены сложней, чем тем соединения, которые принято называть комплексными (K2[PtCl6], [Co(NH3)6]Cl3). Поэтому в рамках данного курса комплексными соединениями будут называться вещества ионного или молекулярного строения, сохраняющие в водных растворах сложные многоатомные группировки, в составе которых центральный атом (комплексообразователь) связан со своими партнерами смешанным ионно-ковалентным взаимодействием, что позволяет ему в зависимости от условий обратимо обменивать своих "соседей" (лиганды).
Чаще всего в качестве комплексообразователя выступают атомы (ионы) d–металлов (например, Fe, Fe2+, Fe3+; Cu+, Cu2+; Ni, Ni2+; Pd2+, Pt2+, Pt4+ и т.п.), ионы р–элементов больших периодов (например, Te4+; Bi3+, Sb3+, Sb5+; Sn2+, Sn4+, Ge4+ и т.п.), а также ионы s–элементов малых периодов (например, Be2+). Даже простое перечисление потенциальных комплексообразователей позволяет заметить, что эта способность наиболее характерна для d–элементов, а наименее характерно комплексообразование для s–элементов. Объясняется это тем, что связь комплексообразователь–лиганд не должна быть ни сильно ионной (иначе такие группировки будут полностью диссоциировать в водном растворе), ни сильно ковалентной (иначе такие группировки будут очень стабильны к замене лигандов, как, например, ионы SO42–). Поэтому центральные атомы должны оказывать среднее поляризующее действие на свое окружение (лиганды).
Таблица 4.7
Систематизация потенциальных
комплексообразователей в зависимости от их положения
в периодической системе и степени окисления
|
Катионы с низким п/д
(образуют сильно ионные связи) |
Катионы со средним п/д (образуют ионно-ковалентные связи)
Потенциальные комплексообразователи |
Катионы с очень высоким п/д
(образуют малополяр-ные кова-лентные связи, по которым не проходит диссоциация, а, значит, невозможен и обмен L) | |||||
|
Li+ Na+
K+
Rb+ Cs+ |
Mg2+
Ca2+
Sr2+ Ba2+ |
Be2+
V2+…Fe2+…Zn2+
Sn2+ Pb2+ |
B3+ Al3+, P3+
Sc3+, Ti3+, V3+, Cr3+, Fe3+, Co3+
Ga3+, As3+ In3+, Sb3+ Tl3+, Bi3+ |
Si4 +
Ti4+,V4+
Ge4+ Zr4+,Sn4+ Hf4+ |
V5+
As5+ Nb5+, Sb5+ Ta5+ |
Mo6+,Te6+ W6+ |
C4+,N3+, N5+
P5+, S4+, S6+ Se4+, Se6+… |
|
La3+ и Ln3+ | |||||||
В некоторых редких случаях к комплексным относят частицы, в составе которых центральным атомом выступает анион (отрицательно поляризованный атом): N–III в ионе аммония [NH4]+, O–II в ионе гидроксония [OH3]+, J–I в полииодид-ионах [JJn]–. Однако это, скорее - дань традиции: большинство особенностей комплексов металлических элементов для приведенных группировок совершенно не характерно.
Как уже отмечалось, связь комплексообразователь–лиганд должна быть в заметной степени ковалентной, а значит должна быть образована за счет обобществления электронов. Потенциальные комплексообразователи на своих валентных орбиталях порой вообще не имеют электронов (например, Be2+, Al3+, Sn4+, Ti4+, V5+). Будучи даже частично ионизированными (например, d–, р–элементы в промежуточных положительных степенях окисления), они обязательно располагают некоторым числом пустых валентных орбиталей. Значит, все они могут выступать акцепторами электронных пар (являются σ–акцепторами). Если число вакантных орбиталей превышает КЧ комплексообразователя, что характерно для атомов (ионов) элементов больших периодов, то они способны проявлять дополнительные π–акцепторные свойства. Такие комплексообразователи характеризуются предпочтительным связыванием с лигандами, имеющими много неподеленных электронных пар (например, с кислород-координирующимися, важнейшими среди которых являются О2–, ОН–, OH2…). d–элементы конца рядов в низких степенях окисления (например, Zn2+, Cu+, Cu2+, Ag+, Au3+, Hg2+), а также р–элементы больших периодов (Sn2+, Sn4+, Sb3+, Ga3+) располагают либо частично (d–элементы), либо полностью заполненными (n-1)d–орбиталями и сами в той или иной степени могут выступать в качестве доноров электронных пар (являются σ–акцепторами и π–донорами). Для них характерно повышенная склонность к связыванию с лигандами, располагающими пустыми валентными орбиталями (Г–, кроме F–; S–координирующимися). Неспаренные электроны, часто имеющиеся на орбиталях d–элементов, могут быть задействованы в σ–связывание с лигандами–радикалами (молекулы NO, NO2).
В качестве лигандов обычно выступают простые одноатомные анионы (Cl–, Br–, J–; реже О2–, S2–), двухатомные анионы (ОН–, SH–, СN–), некоторые многоатомные анионы, не имеющие в своем составе собственного многозарядного центрального атома (SCN–, NO2–, S2O32–, реже SO32–, очень редко NO3–, SO42–), а также полярные (NH3, H2O, CO) и легко поляризуемые молекулы (J2).
Практически все наиболее активно использующиеся потенциальные лиганды способны выступать в качестве доноров электронных пар (σ-доноры). Структуры некоторых из них показаны в табл. 4.8 и на рис. 4.23.
Атомы азота в составе лигандов (аммиак NH3, гидразин N2H4, анионы аминокислот H2NRCOO–) выступают только в качестве σ–доноров (устанавливают только σ–связь за счет собственной электронной пары).
Атомы фтора и кислорода являются и σ–донорами, и π–донорами (особенно хорошо связываются с комплексообразователями, имеющими много пустых валентных орбиталей).
Атомы (ионы) серы, хлора, брома, иода ведут себя как σ–доноры и π–акцепторы (за счет собственных пустых nd–орбиталей). Но особенно сильными π–акцепторными свойствами обладают лиганды, связывающиеся атомами углерода (CN– и CO), т.к. в соответствии с современными теориями химической связи (метод молекулярных орбиталей) в указанных частицах на атомах углерода локализуются вакантные π-разрыхляющие молекулярные орбитали более низколежащие по энергии, чем, например, у атомов S, Г). Соответственно цианидные комплексы и карбонилы более характерны для d–металлов, находящихся во второй половине соответствующих 3d–, 4d– 5d–рядов.
Таблица 4.8
Систематизация потенциальных лигандов
в зависимости от их состава и строения
|
Одноатомные ионы |
Многоатомные ионы |
Молекулы | |
|
|
связываются за счет атомов O, S, C (в полимерных цианидах – и за счет N)
связываются за счет электронных пар атомов S или N N или О
связывается, как правило, атомом S
обычно связывается концевым атомом S |
связывается атомом С | |
|
|
в качестве лигандов выступают крайне редко, т.к. электронные пары атомов кислорода активно участвуют в πp–d–связывании с собственным центральным атомом. |
| |


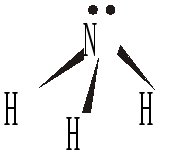

По вполне очевидным причинам простые и водородсодержащие лиганды координируются вокруг комплексообразователя только за счет единственного атома. Такие лиганды могут выступать только в качестве монодентантных ("однозубых"), устанавливают с центральным атомом лишь одну σ–связь. Однако, если атом-донор располагает несколькими электронными парами, то он может использовать их не только для дополнительного π–взаимодействия, но и способен образовать несколько σ–связей с разными атомами металлов, выступить в роли мостикового лиганда в полиядерных островных комплексах или в составе немолекулярных полимерных соединений (см. ниже). Эти же слова применимы и к многоатомным лигандам, содержащим несколько донорных атомов, непосредственно связанных друг с другом (СN–, SCN–, NO2–). По геометрическим причинам они не могут быть одновременно использованы для взаимодействия с одним центральным атомом: это будет равносильно образованию крайне нестабильных трех- или четырехчленных циклов. Поэтому такие лиганды также являются монодентантными, но могут выступать в роли мостиковых. Если же два и больше донорных атома расположены отдаленно друг от друга (через 2-3 других атома в цепи, для этого лиганд должен быть достаточно крупным), то они все могут быть задействованы в σ–связывании с одним комплексообразователем. Такие лиганды (например, этилендиамин NH2CH2CH2NH2, анионы аминоуксусной NH2CH2COO–, щавелевой –OOC–COO– кислот) называют полидентантными ("многозубыми"), хелатообразующими, а соответствующие комплексы – хелатными ("клешнеобразными"). Очень важным лигандом такого типа, дентантность (координационная емкость) которого может меняться от 1 до 6 в зависимости от рН раствора, является анион тетрааминуксусной кислоты (этилен-диамин-тетраацетат (–OOCCH2)2N–CH2-CH2–N(CH2COO–)2, аббревиатура – ЭДТА).

-
Рисунок 4.23 – Структура важных полидентантных лигандов (оксалат-ион, этилендиаминацетат-ион), а также схематичное изображение возможных октаэдрических группировок.
В особую группу принято относить лиганды, не имеющие донорного атома, не устанавливающие с комплексообразователем σ-связей. Такие лиганды, как этилен С2Н4, ацетилен С2Н2,31 бензол С6Н6, циклопентадиенил-ион С5Н5– и некоторые другие способны связываться с d–металлами за счет собственных связывающих π-электронов (в том числе электронов π-систем). Получаемые соединения называют π-комплексами. Есть основания полагать, что таким же способом образуются пероксокомплексы d–металлов.
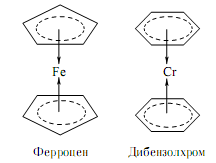
Рис.4.24 Структуры некоторых π-комплексов.